Caso Clínico | Respirar, 2026; 18 (1): 204-211 | ISSN 2953-3414 | https://doi.org/10.55720/respirar.18.1.16
Enfermedades no tan huérfanas: fibroelastosis pleuropulmonar, un desafío diagnóstico
A Not-So-Stray Disease: Pleuroparenchymal Fibroelastosis, a Challenging Diagnosis
Agustina Torres-Vergueiras1 ![]() , Martha I. Rodríguez2
, Martha I. Rodríguez2 ![]() , Santiago Wainer1
, Santiago Wainer1 ![]() ,
,
Luciana Romero2 ![]() , Verónica Torres-Esteche1,2
, Verónica Torres-Esteche1,2 ![]()
1 UdelaR, Facultad de Medicina, Hospital Maciel, Unidad Académica Clínica Médica 3, Montevideo, Uruguay
2 Centro Medica Uruguaya (MUCAM), Departamento de Neumología, Montevideo, Uruguay
Autor corresponsal:
Verónica Torres Esteche. E-mail: torres.esteche@gmail.com
Recibido:
03 junio 2025
Aprobado:
12 octubre 2025
Resumen
La fibroelastosis pleuropulmonar (FEPP) es una enfermedad poco frecuente dentro del espectro de las enfermedades pulmonares intersticiales difusas (EPI). Se clasifica como una neumonía intersticial idiopática rara, según la American Thoracic Society (ATS) y la European Respiratory Society (ERS). Histológicamente, se caracteriza por fibrosis de la pleura visceral asociada a cambios fibroelásticos en el parénquima pulmonar subpleural, con predominio en lóbulos superiores. Su incidencia real es desconocida, probablemente debido al subdiagnóstico y a la ausencia de criterios diagnósticos estandarizados. En este trabajo se reportan dos casos de FEPP en fase avanzada diagnosticados en un centro de Montevideo, Uruguay. Esta entidad representa un desafío diagnóstico y su reconocimiento oportuno es fundamental ya que podría ser más prevalente de lo que se percibe. Debe sospecharse ante pacientes con engrosamiento pleural asociado a compromiso intersticial de lóbulos superiores, lo cual justifica su pronta derivación a centros especializados. Dado su comportamiento progresivo, impacto negativo en la calidad de vida y elevada mortalidad, el tratamiento de la FEPP continúa siendo el principal desafío de esta entidad.
Palabras clave: fibrosis pulmonar; fibroelastosis; enfermedades pulmonares intersticiales difusas.
Abstract
Pleuroparenchymal fibroelastosis (PPFE) is a rare disease within the spectrum of diffuse interstitial lung diseases, classified as a rare idiopathic interstitial pneumonia by the American Thoracic Society (ATS) and the European Respiratory Society (ERS). Histologically, it is characterized by fibrosis of the visceral pleura associated with fibroelastic changes in the subpleural lung parenchyma, predominantly in the upper lobes. Its real incidence is unknown, due to underdiagnosis and the lack of standardized diagnostic criteria. This article reports two clinical cases diagnosed with advanced PPFE in a medical center in Montevideo, Uruguay. This condition presents a diagnostic challenge, and its early recognition is crucial as it might be more prevalent than perceived. PPFE should be suspected in patients with pleural thickening associated with upper lobes interstitial involvement, which justifies prompt referral to specialized centers. Given its progressive behavior, negative impact on quality of life, and high mortality, PPFE treatment remains the main challenge for this entity.
Keywords: pulmonary fibrosis; fibroelastosis; interstitial lung diseases.
Introducción
La fibroelastosis pleuropulmonar (FEPP) es una entidad infrecuente, del grupo de las enfermedades pulmonares intersticiales difusas (EPI). Se clasifica dentro de las neumonías intersticiales idiopáticas (NII) raras, según la American Thoracic Society y European Respiratory Society.1,2
Su histología combina fibrosis de pleura visceral y cambios fibroelásticos en el parénquima pulmonar subpleural, con predilección por los lóbulos superiores.
La incidencia de FEPP es incierta, atribuible al subdiagnóstico y la falta de criterios diagnósticos uniformes.3,4
Estudios reportan una prevalencia del 7,7% de FEPP dentro de las NII en un período de 10 años.5 Clínicamente, su evolución puede ser similar a la fibrosis pulmonar idiopática (FPI).6
Se han postulado factores desencadenantes como infecciones respiratorias previas,7-12 mientras que no se ha encontrado asociación significativa con tabaquismo o estados de inmunosupresión específicos.13
Casos clínicos
Caso 1
Hombre de 63 años, cocinero con exposición a biomasa, ex tabaquista, sin antecedentes de tuberculosis, ni enfermedades autoinmunes, ni exposición a fármacos neumotóxicos. Relataba disnea progresiva de 1 año de evolución y descenso de peso.
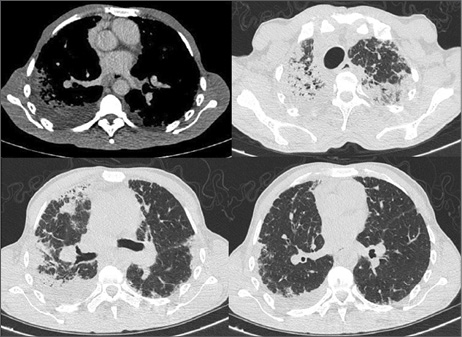
Ingresó por neumonía. Tomografía (TC) de tórax (Figura 1) mostró disminución volumétrica de lóbulo superior derecho, con engrosamiento intersticial, consolidaciones y bronquiectasias por tracción en ambos lóbulos superiores, compromiso intersticial en lóbulos inferiores, derrame pleural derecho. Lavado bronquiolo-alveolar: Pseudomonas Aeruginosa; se descartó tuberculosis y hongos. Resuelto el cuadro infeccioso, mantuvo disnea y crepitantes tipo velcro.
Figura 1.
Caso 1. TC inicial. Disminución volumétrica de lóbulos superiores a predominio derecho. Engrosamiento del intersticio bilateral de tipo reticular, a predominio subpleural, asociado a consolidaciones y bronquiectasias por tracción en lóbulos superiores. Derrame pleural derecho.
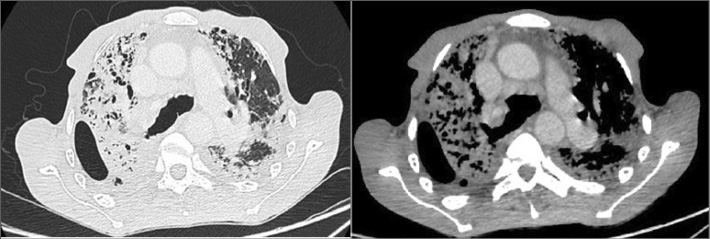
TC de control (Figura 2): neumotórax y progresión del compromiso intersticial. Espirometría: patrón restrictivo severo, capacidad vital forzada (CVF): 35%.
Figura 2
Caso 1. TC en evolución. Progresión de los cambios fibróticos, neumotórax derecho de moderada entidad, con engrosamiento pleural.
Biopsia transbronquial mostró mucosa con epitelio cilíndrico respiratorio, tejido conectivo fibroso hialinizado, infiltrado linfomonocitario. Sin granulomas ni microorganismos. (Figuras 3 y 4)
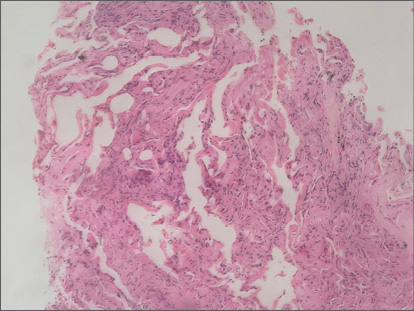
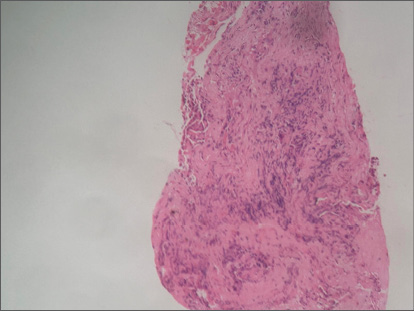
Figura 3.
Caso 1 Anatomía patológica. Biopsia transbronquial: tejido conectivo fibroso, hialinizado.
Figura 4.
Caso 1 Anatomía patológica. Biopsia transbronquial: importante infiltrado linfomonocitario.
Anticuerpos antinucleares y anti nucleocitoplasmáticos negativos; proteinograma electroforético e inmunoglobulinas normales. Fibroesofagogastroscopia normal. Se descartaron neoplasias.
En el ateneo de EPI, se planteó diagnóstico de FEPP. La evolución fue desfavorable y no fue candidato a trasplante pulmonar.
Caso 2
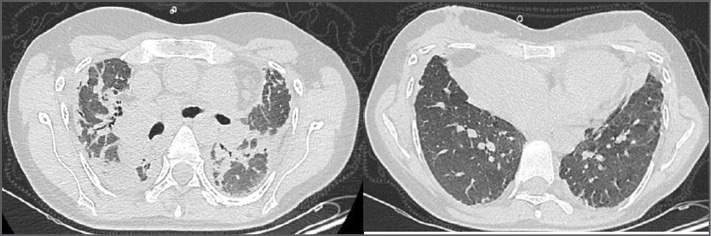
Mujer de 39 años con exposición a humo de biomasa durante 3 años, sin antecedentes de tabaquismo, nuemotóxicos, tuberculosis, ni enfermedades autoinmunes. Presentó dolor torácico, disnea y pérdida de peso de 4 años de evolución. Fue inicialmente diagnosticada con secuelas de tuberculosis. (Figura 5) Una nueva TC evidenció opacidades fibro-retráctiles pleuropulmonares, disminución volumétrica de lóbulos superiores, distorsión arquitectural con progresión hacia lóbulos inferiores y marcada depresión en horquilla supraesternal. (Figura 6) Patrón restrictivo muy severo (CVF 23%), difusión de monóxido de carbono (DLCO), descenso severo 25%. Biopsia pleural, pericárdica y pulmonar mostraron: neumonitis intersticial-septal con predominio de fibrosis, sin patrón de neumonía intersticial usual, ni granulomas ni microorganismos; además pleuritis y pericarditis crónicas inespecíficas.
Figura 5
Caso 2. TC de tórax inicial. Engrosamientos pleurales apicales, disminución volumétrica de lóbulos superiores, consolidaciones pulmonares fibro-retráctiles, con distorsión de arquitectura pulmonar y extensión a lóbulos inferiores. Marcada depresión en la horquilla supraesternal.
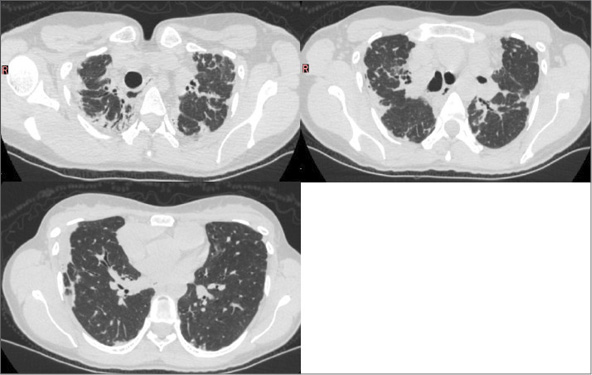
Figura 6.
Caso 2. TC de Tórax en evolución. Progresión del compromiso fibro-retráctil del parénquima pulmonar, a predominio de lóbulos superiores.
En ateneo multidisciplinario, se planteó FEPP. Se descartó patología autoinmune. No fue candidata a trasplante pulmonar, debido a la alta morbimortalidad relacionada con adherencias pleurales. Falleció a los 2 años del diagnóstico.
Discusión
La FEPP puede ser idiopática, como en los casos presentados, o secundaria; pudiendo asociarse a trasplante, radioterapia, quimioterapia, enfermedades autoinmunes sistémicas (EAS), neumonitis por hipersensibilidad fibrótica y a otras EPI, incluyendo FPI.14
Entre las EAS más relacionadas, se encuentran la esclerosis sistémica, artritis reumatoidea, síndrome de Sjögren y enfermedad inflamatoria intestinal.8,15,16
Bonifazi et al. encontraron FEPP en 18% de pacientes con esclerosis sistémica (n=359), con peor supervivencia independientemente de la gravedad de la enfermedad.17 Kang et al. reportaron un 6,5% de FEPP en 477 pacientes con artritis reumatoidea y enfermedad pulmonar intersticial difusa.18
La FEPP puede ocurrir en formas familiares de neumonitis intersticiales idiopáticas, principalmente en mujeres jóvenes. La paciente joven del caso 2 no tenía antecedentes familiares. La mayoría de los casos de FEPP se da en pacientes no fumadores.19
La fisiopatología es incierta, pero el espectro clínico y comportamiento de la enfermedad sugieren que es resultado de la combinación de predisposición genética, desregulación del sistema inmune y exposición ambiental.
Clínicamente, la FEPP se manifiesta con tos, disnea y pérdida de peso progresiva con bajo peso marcado en etapas avanzadas, como en los casos presentados. La edad de inicio es variable.
El neumotórax ocurre en aproximadamente un tercio de los pacientes (caso 1) y el dolor pleurítico puede estar presente aún sin neumotórax (caso 2).1
Del examen físico, se destaca la depresión del hueco supraesternal, como se observaba en ambos casos.
En estadios avanzados puede aparecer platitórax o “tórax plano”, caracterizado por reducción del diámetro anteroposterior torácico y respecto al transversal, secundario a la disminución volumétrica de lóbulos superiores, lo que contribuye al patrón restrictivo observado.6,20,21 El caso 2 presentaba platitórax.
El diagnóstico se plantea ante hallazgos imagenológicos característicos, como en los casos analizados.3 Se describen los hallazgos imagenológicos más frecuentes en la Tabla 1. En un subgrupo de pacientes se ha demostrado extensión a los lóbulos inferiores con patrón de afectación de neumonitis intersticial usual.22 Una serie de pacientes (n= 21) informó una afectación de los lóbulos superiores e inferiores en un tercio de los casos.21,23
Tabla 1.
Hallazgos imagenológicos en FEPP. Adaptada de Chua F, et al ( 2019).3
|
Engrosamiento pleural y fibrosis subpleural en lóbulos superiores |
Pérdida de volumen pulmonar en lóbulos superiores |
|
Hilos retraídos con distorsión de la arquitectura pulmonar |
Relación reducida entre el diámetro torácico anteroposterior y el transversal |
|
Demarcación clara entre pulmón anormal y normal |
Focos de consolidación, quistes, engrosamiento intersticial interlobulillar, neumotórax |
Los principales diagnósticos diferenciales de FEPP se plantean con patologías que predominan en lóbulos superiores: neumonitis por hipersensibilidad (HP), sarcoidosis, NII con extensión a las zonas superiores (incluyendo NIU), infección micobacteriana no tuberculosa, remodelación posterior a lesión pulmonar, neumoconiosis y malignidad.3 En los casos analizados, se descartaron estas entidades, por lo cual se plantearon como FEPP idiopáticas.
La biopsia quirúrgica presenta significativo riesgo de complicaciones como neumotórax o fístula broncopleural.24 En algunas series, la FEPP se diagnostica frecuentemente por discusión multidisciplinaria (MDD) sin necesidad de biopsia. En el trabajo de Nasser et al., el diagnóstico de FEPP se confirmó en MDD, obviando la necesidad de biopsia en 16 de 21 casos.25
Histológicamente, se caracteriza por engrosamiento fibroso de la pleura visceral, fibrosis intraalveolar homogénea con elastosis septal, transición abrupta entre tejido afectado y normal, e infiltración linfocitaria.1,26,27
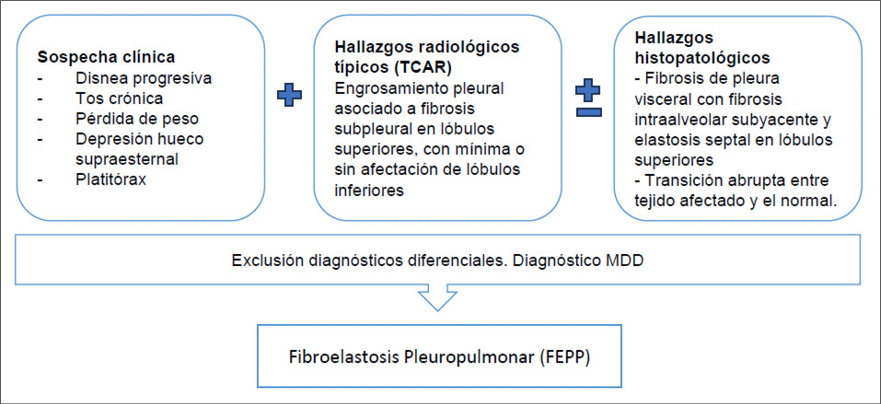
En ambos casos presentados, existió un retraso diagnóstico, realizándose diferentes planteos al inicio de la enfermedad, lo cual coincide con la literatura. Esto puede deberse a que es una enfermedad rara, con larga fase subclínica y cuyo diagnóstico obliga a descartar otras entidades. Si bien no existen criterios diagnósticos estandarizados, se plantean criterios clínicos, imagenológicos e histopatológicos. (Figura 7)
Figura 7.
Criterios diagnósticos de FEPP. Adaptada de Chua F, et al. (2019) 3
En cuanto al tratamiento y manejo, se destaca que no existe ningún tratamiento validado para la FEPP. La prednisolona en dosis bajas podría tener efectos inmunomoduladores útiles. El uso de dosis más altas de corticosteroides o de inmunosupresores como azatioprina o metotrexato generalmente se evita por el mayor riesgo de infecciones.5,25
La experiencia con antifibróticos es escasa en FEPP y no hay evidencia sólida para esta enfermedad; sin embargo, los mismos retardan la progresión de la enfermedad en pacientes con EPI fibrótica de otras entidades, como la FPI, la EPI asociada a esclerosis sistémica, la EPI con fenotipo de fibrosis progresiva y la EPI progresiva idiopática no clasificable.28-31 La Pirfenidona fue utilizada en casos aislados, con resultados variables.5,32Nasser et al. analizaron la respuesta a Nintedanib en 5 pacientes con FEPP y concluyeron que el tratamiento podría ser beneficioso.25
Por otro lado, es fundamental implementar medidas generales que mejoren la calidad de vida y el pronóstico, tales como: vacunación antineumocócica y contra influenza, rehabilitación pulmonar, oxigenoterapia en caso de hipoxemia y manejo adecuado de comorbilidades. La rehabilitación respiratoria no optimiza la capacidad funcional, sino que mejora la calidad de vida, la tolerancia al ejercicio y la disnea.
El trasplante pulmonar es la única opción potencialmente curativa y se han descrito casos exitosos, pero pueden existir limitaciones técnicas importantes por el engrosamiento pleural extenso y presenta alta morbimortalidad asociada. 33-36
Las principales causas de muerte son la progresión de la enfermedad e infecciones.37 Algunos pacientes siguen un curso progresivo con insuficiencia respiratoria irreversible y muerte temprana.3 Sin embargo, la progresión de FEPP es variable: algunos pacientes muestran una progresión rápida, pero otros tienen progresión lenta de 10 a 20 años.21,23
La supervivencia media de nuestros pacientes fue de 8 años. Algunos factores pueden contribuir a una peor evolución, como patrón NIU asociado o formas familiares.13,14
Conclusiones
La fibroelastosis pleuropulmonar es una entidad subdiagnosticada que debe considerarse en el diagnóstico diferencial de fibrosis pulmonar apical bilateral.38 Su reconocimiento temprano es fundamental para la derivación a centros especializados. El curso clínico es progresivo y afecta negativamente la calidad de vida, con alta mortalidad y opciones terapéuticas limitadas. Creemos que este trabajo aporta al conocimiento de esta entidad y sería de interés contar con registros nacionales y estudios multicéntricos que permitan profundizar en su manejo.
Financiamiento: los autores declaran que el trabajo no tuvo financiamiento.
Conflictos de interés: los autores declaran que no tienen conflictos de intereses relacionados con el tema de esta publicación.
Contribuciones de los autores: ATV: revisión y edición, curaduría de datos, escritura. MIR: análisis formal, redacción, supervisión. SW: revisión y edición, curaduría de datos, escritura. LR: revisión, investigación. VTE: administración del proyecto, análisis formal, redacción, supervisión.
Los Editores en Jefe, Dres. Carlos Luna y Francisco Arancibia, realizaron el seguimiento del proceso de revisión y aprobaron este artículo.
Referencias
1. Bonifazi M, Montero MA, Renzoni EA. Idiopathic Pleuroparenchymal Fibroelastosis. Curr Pulmonol Reports 2017;6(1):9–15. https://doi.org/10.1007/s13665-017-0160-5
2. Giménez A, Mazzini S, Franquet T. El informe radiológico en patología intersticial pulmonar. Radiologia 2022;64:142–9. https://doi.org/10.1016/j.rx.2022.01.007
3. Chua F, Desai SR, Nicholson AG, Devaraj A, Renzoni E, Rice A et al. Pleuroparenchymal fibroelastosis a review of clinical, radiological, and pathological characteristics. Ann Am Thorac Soc 2019;16(11):1351–9. https://doi.org/10.1513/AnnalsATS.201902-181CME
4. Frankel SK, Cool CD, Lynch DA, Brown KK. Idiopathic pleuroparenchymal fibroelastosis: description of a novel clinicopathologic entity. Chest 2004;126(6):2007–2013. https://doi.org/10.1378/chest.126.6.2007
5. Shioya M, Otsuka M, Yamada G, Umeda Y, Ikeda K, Nishikiori H et al. Poorer prognosis of idiopathic pleuroparenchymal fibroelastosis compared with idiopathic pulmonary fibrosis in advanced stage. Can Respir J 2018;2018:6043053. https://doi.org/10.1155/2018/6043053
6. Ishii H, Kinoshita Y, Kushima H, Nagata N, Watanabe K. The similarities and differences between pleuroparenchymal fibroelastosis and idiopathic pulmonary fibrosis. Chron Respir Dis 2019;16:1–9. https://doi.org/10.1177/1479973119867945
7. Beynat-Mouterde C, Beltramo G, Lezmi G, Pernet D, Camus C, Fanton A et al. Pleuroparenchymal fibroelastosis as a late complication of chemotherapy agents. Eur Respir J 2014;44:523–527. https://doi.org/10.1183/09031936.00214713
8. Enomoto Y, Nakamura Y, Colby TV, Johkoh T, Sumikawa H, Nishimoto K et al. Radiologic pleuroparenchymal fibroelastosis-like lesion in connective tissue disease-related interstitial lung disease. PLoS One 2017;12:e0180283. https://doi.org/10.1371/journal.pone.0180283
9. Jacob J, Odink A, Brun AL, Macaluso C, de Lauretis A, Kokosi M et al. Functional associations of pleuroparenchymal fibroelastosis and emphysema with hypersensitivity pneumonitis. Respir Med 2018;138:95–101. https://doi.org/10.1016/j.rmed.2018.03.031
10. Watanabe K. Pleuroparenchymal fibroelastosis: its clinical characteristics. Curr Respir Med Rev 2013;9:299–237. https://doi.org/10.2174/1573398X0904140129125307
11. Xu L, Rassaei N, Caruso C. Pleuroparenchymal fibroelastosis with long history of asbestos and silicon exposure. Int J Surg Pathol 2018;26:190–193. https://doi.org/10.1177/1066896917739399
12. Piciucchi S, Tomassetti S, Casoni G, Sverzellati N, Carloni A, Dubini A et al. High resolution CT and histological findings in idiopathic pleuroparenchymal fibroelastosis: features and differential diagnosis. Respir Res 2011;12:111–115. https://doi.org/10.1186/1465-9921-12-111
13. Von der Thüsen JH. Pleuroparenchymal fibroelastosis: its pathological characteristics. Curr Respir Med Rev 2013;9:238–247.https://doi.org/10.2174/1573398X113096660025
14. Ricoy J, Suárez-Antelo J, Antúnez J, Martínez de Alegría A, Ferreiro L, Toubes ME et al. Pleuroparenchymal fibroelastosis: Clinical, radiological and histopathological features. Respir Med 2022;191:1–7. https://doi.org/10.1016/j.rmed.2021.106437
15. Reddy TL, Tominaga M, Hansell DM, Von Der Thusen J, Rassl D, Parfrey H et al. Pleuroparenchymal fibroelastosis: A spectrum of histopathological and imaging phenotypes. Eur Respir J 2012;40(2):377–85. https://doi.org/10.1183/09031936.00165111
16. Morán Álvarez P, Bachiller-Corral J, Gorospe Sarasúa L, de la Puente Bujidos C. Fibroelastosis pleuroparenquimatosa: un nuevo tipo de neumonía intersticial asociada a conectivopatías. Reumatol Clínica 2020;16(6):513–4. https://doi.org/10.1016/j.reuma.2018.09.003
17. Bonifaci M, Sverzellati N, Negri E, Jacob J, Egashira R, Moser J et al. Pleurparenchymal fibroelastosis in systemic sclerosis: prevalence and prognostic impact. Eur Respir J 2020;56:1902135. https://doi.org/10.1183/13993003.02135-2019
18. Kang J, Seo WJ, Lee EY, Chang SH, Choe J, Hong S et al. Pleuroparenchymal fibroelastosis in rheumatoid arthritis-associated interstitial lung disease. Respir Res 2022;23(1):1–10. https://doi.org/10.1186/s12931-022-02064-z
19. Kokosi MA, Nicholson AG, Hansell DM, Wells AU. Rare idiopathic interstitial pneumonias: LIP and PPFE and rare histologic patterns of interstitial pneumonias: AFOP and BPIP. Respirology 2016;21(4):600–14 https://doi.org/10.1111/resp.12693
20. Harada T, Yoshida Y, Kitasato Y, Tsuruta M, Wakamatsu K, Hirota T et al. The thoracic cage becomes flattened in the progression of pleuroparenchymal fibroelastosis. Eur Respir Rev 2014;23(132):263–6. https://doi.org/10.1183/09059180.00006713
21. Ishii H, Watanabea K, Kushimaa H, Babab T, Watanabec S, Yamadad Y. Pleuroparenchymal fibroelastosis diagnosed by multidisciplinary discussions in Japan. Resp Med 2018;141:190-197. https://doi.org/10.1016/j.rmed.2018.06.022">https://doi.org/10.1016/j.rmed.2018.06.022
22. Rosenbaum JN, Butt YM, Johnson KA, Meyer K, Batra K, Kanne JP et al. Pleuroparenchymal fibroelastosis: a pattern of chronic lung injury. Hum Pathol 2015;46(1):137–46. https://doi.org/10.1016/j.humpath.2014.10.007
23. Yoshida Y, Nagata N, Tsuruta N, Kitasato Y, Wakamatsu K, Yoshimi M et al. Heterogeneous clinical features in patients with pulmonary fibrosis showing histology of pleuroparenchymal fibroelastosis. Respir Investig 2016;54(3):162–9. https://doi.org/10.1016/j.resinv.2015.11.002
24. Becker CD, Gil J, Padilla ML. Idiopathic pleuroparenchymal fibroelastosis: an unrecognized or misdiagnosed entity? Mod Pathol 2008;21:784–787. https://doi.org/10.1038/modpathol.2008.56
25. Nasser M, Si-Mohamed S, Turquier S, Traclet J, Ahmad K, Philit F et al. Nintedanib in idiopathic and secondary pleuroparenchymal fibroelastosis. Orphanet J Rare Dis 2021;16(1):1–9. https://doi.org/10.1186/s13023-021-02043-5
26. Kinoshita Y, Ishii H, Nabeshima K, Watanabe K. The pathogenesis and pathology of idiopathic pleuroparenchymal fibroelastosis. Histol Histopathol 2021;36(3):291–303. https://doi.org/10.14670/HH-18-289
27. Kusagaya H, Nakamura Y, Kono M, Kaida Y, Kuroishi S. Idiopathic pleuroparenchymal fibroelastosis: consideration of a clinicopathological entity in a series of Japanese patients. BMC Pulmonary Medicine 2012;12:72. https://doi.org/10.1186/1471-2466-12-72
28. Richeldi L, Du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U et al. Efcacy and safety of nintedanib in idiopathic pulmonary fbrosis. N Eng J Med 2014;370:2071–82. https://doi.org/10.1056/NEJMoa1402584
29. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Eng J Med 2019;380:2518–28. https://doi.org/10.1056/NEJMoa1903076
30. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Eng J Med 2019;381:1718–27. https://doi.org/10.1056/NEJMoa1908681
31. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med 2020;8:147–57. https://doi.org/10.1016/S2213-2600(19)30341-8
32. Sato S, Hanibuchi M, Takahashi M, Fukuda Y, Morizumi S, Toyoda Y et al. A patient with idiopathic pleuroparenchymal fibroelastosis showing a sustained pulmonary function due to treatment with pirfenidone. Intern Med 2016;55:497–501. https://doi.org/10.2169/internalmedicine.55.5047
33. Chen F, Matsubara K, Miyagawa-Hayashino A, Tada K, Handa T, Yamada T et al. Lung transplantation for pleuroparenchymal fibroelastosis after chemotherapy. Ann Thorac Surg 2014;98:e115–e117. https://doi.org/10.1016/j.athoracsur.2014.07.045
34. Righi I, Morlacchi L, Rossetti V, Mendogni P, Palleschi A, Tosi D et al. Lung transplantation as successful treatment of end-stage idiopathic pleuroparenchymal fibroelastosis: a case report. Transplant Proc 2019;51:235–238. https://doi.org/10.1016/j.transproceed.2018.04.071
35. Hata A, Nakajima T, Yoshida S, Kinoshita T, Terada J, Tatsumi K et al. Living donor lung transplantation for pleuroparenchymal fibroelastosis. Ann Thorac Surg 2016;101:1970–1972. https://doi.org/10.1016/j.athoracsur.2015.07.056
36. Ali MS, Ramalingam VS, Haasler G, Presberg K. Pleuroparenchymal fibroelastosis (PPFE) treated with lung transplantation and review of the literature. BMJ Case Rep 2019;12(4). https://doi.org/10.1136/bcr-2019-229402
37. Cottin V, Si-Mohamed S, Diesler R, Bonniaud P, Valenzuela C. Pleuroparenchymal fibroelastosis. Curr Opin Pulm Med 2022;28(5):432–40. https://doi.org/10.1097/MCP.0000000000000907
38. Portillo K, Guasch Arriaga I, Ruiz-Manzano J. Fibroelastosis pleuropulmonar: ¿es también una entidad idiopática? Arch Bronconeumol 2015;51(10):509–14. https://doi.org/10.1016/j.arbres.2015.05.002
 Esta revista está bajo una licencia de Creative Commons Attribution 4.0 International
Esta revista está bajo una licencia de Creative Commons Attribution 4.0 International