Revisión | Respirar, 2026; 18 (1): 185-198 | ISSN 2953-3414 | https://doi.org/10.55720/respirar.18.1.14
Fisiopatología y relevancia clínica de los monocitos en la fibrosis pulmonar idiopática: revisión narrativa
Pathophysiology and Clinical Relevance of Monocytes in Idiopathic Pulmonary Fibrosis: a Narrative Review
Juan Pablo Camargo Mendoza ![]()
Hospital Central de la Policía Nacional y O&C, Unidad de Neumología, Bogotá, Colombia
AUTOR CORRESPONSAL:
Dr. Camargo Mendoza. E-mail: jpcamargome@unal.edu.co
Recibido:
febrero 2025
Aprobado:
7 noviembre 2025
Resumen
Las enfermedades pulmonares intersticiales son un grupo heterogéneo de desórdenes caracterizados por grado variable de inflamación y fibrosis. La fibrosis pulmonar idiopática (FPI) es una de las más frecuentes y se relaciona con pobre pronóstico, para lo cual se han desarrollado múltiples biomarcadores para predecir de forma temprana el curso clínico y respuesta al manejo. Los monocitos se han relacionado en la patogénesis de la enfermedad fibrótica aberrantemente. La proliferación de precursores de monocitos en la médula ósea y la posterior migración de monocitos a la sangre y los tejidos es un proceso estrechamente relacionado con los macrófagos alveolares. CCR2 es el receptor necesario para la migración clásica de monocitos a tejidos inflamados. Entre las múltiples quimiocinas asociadas, se destaca la proteína quimioatrayente de monocitos 1 (MCP-1, también conocida como CCL2) que tiene la mayor afinidad por CCR2. Se han encontrado niveles de monocitos activados en pacientes con FPI y los monocitos circulantes pueden producir proteínas de la matriz celular profibróticas. El conteo elevado de monocitos en sangre se ha relacionado con progresión y con incremento de la probabilidad de muerte en FPI.
Palabras clave: monocito; fibrosis pulmonar idiopática; biomarcador; pronóstico.
Abstract
Interstitial lung diseases are a heterogeneous group of disorders characterized by variable degrees of inflammation and fibrosis. Idiopathic pulmonary fibrosis (IPF) is one of the most common types of lung fibrosis and is linked to a poor prognosis. Several biomarkers have been developed to help doctors predict early on the disease’s course and how well it will respond to treatment. The pathogenesis of fibrotic disease aberrantly implicates monocytes. This process, where monocyte precursors grow in the bone marrow and monocytes move to other tissues, is a lot like alveolar macrophages. CCR2 is the receptor necessary for the classical migration of monocytes to inflamed tissues. This chemokine, monocyte chemoattractant protein 1 (MCP-1, also called CCL2), binds to CCR2 better than any other. Activated monocytes have been found in IPF patients. Patients with activated monocytes can produce profibrotic proteins. Research has linked high blood monocyte counts to the progression of IPF and an increased risk of death.
Keywords: monocyte; idiopathic pulmonary fibrosis; biomarker; prognosis.
Introducción
La fibrosis pulmonar idiopática (FPI) constituye aproximadamente el 30% de todas las enfermedades intersticiales difusas (EPID); su incidencia en adultos es 3 a 9 por cada 100.000, siendo más frecuente en hombres que mujeres en una razón 3:1. La FPI es una enfermedad pulmonar crónica, progresiva y de etiología multifactorial, caracterizada por una remodelación fibrótica del parénquima pulmonar que conduce a un deterioro irreversible de la función respiratoria. A pesar de los avances terapéuticos, la FPI mantiene un pronóstico reservado, con una mediana de supervivencia de 2 a 5 años tras el diagnóstico.1,2
La evolución clínica de la FPI es heterogénea, lo que dificulta la predicción del curso de la enfermedad y la toma de decisiones terapéuticas. En este contexto, la identificación de biomarcadores fiables y accesibles es crucial para mejorar la estratificación de pacientes, monitorear la progresión y evaluar la respuesta al tratamiento.3,4
Entre los biomarcadores en FPI diferencialmente expresados destacan KL-6, SP-A y SP-D, relacionados con la disfunción de las células epiteliales alveolares (CEA) y la inflamación persistente en el microambiente pulmonar.15 Además, enzimas como MMP-1, MMP-7 y MMP-28 se vinculan a la remodelación de la MEC, mientras que proteínas como POSTN y osteopontina (OPN) emergen como reguladores clave en la progresión.4 La diferenciación entre FPI y otras EPID representa un desafío clínico, donde biomarcadores como MMP-28 sérico, POSTN y OPN han demostrado valor en la discriminación de FPI frente a entidades como la neumonitis por hipersensibilidad crónica (NHC) y la neumonía intersticial no específica (NINE). Estos biomarcadores complementan el arsenal diagnóstico y facilitan la caracterización precisa de los pacientes.16
Durante las exacerbaciones agudas, se observa una intensificación de la respuesta inflamatoria y fibrótica. Biomarcadores como S100A9, α-defensina, CXCL8, CXCL13, CCL2, IL-6, LTBP2 y HMGB1 muestran incrementos significativos, y reflejan el estado de activación inmune y el daño alveolar.17-19 La progresión de la FPI implica un deterioro funcional continuo y mayor riesgo de mortalidad. En este contexto, biomarcadores como KL-6, SP-A, SP-D, RAGE, MMP-7, POSTN, YKL-40 y monocitos han mostrado asociación con la disminución de la función pulmonar y la progresión.20,21 La severidad de la FPI puede correlacionarse con biomarcadores que reflejan tanto la carga fibrótica como la respuesta inflamatoria. Entre ellos, se destacan KL-6, RAGE, SAA, CEA, VEGF, MMP-7, YKL-40 y los monocitos.22 ,23.
Algunos trabajos han evaluado a los monocitos circulantes como posibles biomarcadores en FPI. Se ha demostrado que un recuento elevado de monocitos en sangre periférica se asocia con una mayor tasa de progresión de la enfermedad, hospitalizaciones y mortalidad en pacientes con FPI.7,8 Además, análisis transcriptómicos han revelado que los monocitos en FPI presentan una activación aberrante de vías inmunológicas, que podría contribuir a la patogénesis de la fibrosis.8 Dado que el recuento de monocitos es una medida sencilla, económica y ampliamente disponible en la práctica clínica, su potencial como biomarcador pronóstico en FPI es particularmente atractivo. En esta revisión, se profundizará en el papel de los monocitos como biomarcadores en la FPI, explorando su relevancia en la patogenia, su potencial utilidad clínica en el diagnóstico y seguimiento de la enfermedad, y las perspectivas futuras para su aplicación.
Metodología
Se realizó una búsqueda bibliográfica estructurada en PubMed/MEDLINE, Embase, Web of Science, ScienceDirect y Cochrane Library sobre publicaciones comprendidas entre los años 2000 y 2024, en inglés. Para PubMed se emplearon términos MeSH y sus equivalentes, combinados con operadores booleanos: (“Idiopathic Pulmonary Fibrosis”[MeSH] OR “Pulmonary Fibrosis, Idiopathic”) AND (“Monocytes”[MeSH] OR “Blood Monocyte Count”) AND (“Biomarkers”[MeSH] OR “Prognosis”[MeSH] OR “Disease Progression”) AND (“Acute Exacerbation” OR “Exacerbations, Pulmonary”) AND (“Antifibrotic Agents”[MeSH] OR “Pirfenidone” OR “Nintedanib”). La depuración de los resultados se realizó en fases: en la primera, se eliminaron artículos duplicados; posteriormente, se seleccionaron aquellos relacionados con la fisiopatología de la FPI y la identificación general de biomarcadores; finalmente, se restringió la búsqueda a la evidencia específica sobre el papel de los monocitos en la FPI, incluyendo su relación con exacerbaciones agudas y respuesta al tratamiento antifibrótico. De un total de 168 artículos identificados inicialmente, se seleccionaron 58 estudios para la revisión cualitativa final.
Bases fisiopatogénicas generales de la FPI
La FPI se produce como resultado de una lesión epitelial y una desregulación de la interacción epitelio/ mesénquima, que activa continuamente múltiples vías profibróticas interconectadas, lo que en última instancia conduce a una respuesta de reparación anormal y a la pérdida de la función pulmonar.9
En el alvéolo sano, la membrana basal íntegra asegura el intercambio gaseoso. Tras la agresión, las células epiteliales alveolares dañadas y los capilares liberan citocinas inflamatorias (IL-1β, IL-17A, IL-6, IL-10, IL-12) y factores de coagulación, lo que genera un microambiente inflamatorio. La transición epitelio-mesenquimatosa (TEM) y el reclutamiento de progenitores mesenquimales y fibrocitos inducen la formación de focos fibroblásticos.10 Allí, los fibroblastos se diferencian en miofibroblastos, células clave en la síntesis y depósito de matriz extracelular (MEC). Bajo el estímulo del factor de crecimiento transformante beta (TGF-β), la acumulación de MEC se vuelve progresiva, lo que produce rigidez pulmonar y deterioro de la distensibilidad y del intercambio gaseoso.12,13
Paralelamente, la inmunidad adaptativa contribuye a la perpetuación del daño. Se han identificado infiltrados de linfocitos T y B, y la presencia de estructuras linfoides terciarias en tejido pulmonar y lavado broncoalveolar, lo que sugiere que incluso en áreas aparentemente “inactivas” persiste una señal inmunológica activa.14
En este entramado, los monocitos ocupan un lugar central. Desde la circulación periférica, son reclutados por quimiocinas como CCL2/CCR2 y migran al pulmón, donde se diferencian en macrófagos intersticiales. Estos no solo mantienen la inflamación mediante secreción de citocinas, sino que también potencian la activación del eje TGF-β–miofibroblasto, lo que facilita el remodelado patológico. La hipoxia presente en el microambiente fibrótico refuerza este reclutamiento y diferenciación, lo que intensifica la producción de mediadores profibróticos.8
Biomarcadores en FPI
Un biomarcador se define como una característica que se mide y evalúa objetivamente como indicador de procesos biológicos normales, procesos patógenos o respuestas farmacológicas a una intervención terapéutica.58
En la FPI, los biomarcadores constituyen indicadores objetivos y reproducibles que abarcan desde parámetros clínico-funcionales (CVF, DLCO) y hallazgos radiológicos, hasta determinantes moleculares y celulares derivados de tejido pulmonar, lavado broncoalveolar y sangre periférica, con valor diagnóstico, pronóstico y terapéutico.3 (Tabla 1) Entre los más relevantes destacan KL-6, SP-A y SP-D como reflejo de disfunción epitelial alveolar; MMP-1, MMP-7 y MMP-28 vinculados a remodelado de matriz extracelular; y POSTN y osteopontina como moduladores clave de progresión fibrótica, útiles además para diferenciar FPI de otras enfermedades intersticiales difusas.4,15,16 Durante las exacerbaciones agudas, biomarcadores inflamatorios como S100A9, α-defensinas, CXCL8, CXCL13, CCL2, IL-6, LTBP2 y HMGB1 evidencian la intensificación del daño alveolar y la activación inmune. En el seguimiento longitudinal, marcadores como KL-6, RAGE, MMP-7, YKL-40 y, de forma destacada, los monocitos, se asocian con deterioro funcional acelerado, mayor riesgo de exacerbaciones y mortalidad, consolidándose estos últimos como un biomarcador celular simple, económico y de alto valor pronóstico.17,19,20,21 En la evaluación terapéutica, IGFBP-2 sérico, SP-D y Gal-3 han demostrado utilidad al reflejar la eficacia de antifibróticos como pirfenidona y nintedanib, consolidando un marco multidimensional en el que la integración de biomarcadores epiteliales, de remodelado, inflamatorios y celulares permite caracterizar la enfermedad, estratificar riesgo y guiar estrategias de medicina personalizada en FPI.24,26 (Tabla 2)
Tabla 1.
Clasificación estructurada de biomarcadores reportados en FPI.
|
Tipo de biomarcador |
Ejemplos destacados |
Técnica de detección / Fuente |
Aplicación clínica principal |
|---|---|---|---|
|
Bioquímico |
KL-6, SP-A, SP-D, MMP-7, periostina |
Suero, plasma, LBA / ELISA, inmunoensayo |
Diagnóstico, pronóstico, monitoreo de progresión |
|
Radiológico |
Patrón NIU, NIU probable, NIU indeterminada |
TCAR |
Categorización diagnóstica, evaluación de extensión |
|
Funcional pulmonar |
CVF, DLCO |
Pruebas de función pulmonar (espirometría, difusión) |
Estratificación de severidad, seguimiento longitudinal |
|
Genético |
Polimorfismo MUC5B, TERT, TERC |
Análisis de SNP, qPCR, secuenciación (SNG) |
Predicción de susceptibilidad, progresión |
|
Transcriptómico |
Firma génica de 52 genes, perfiles de monocíticos |
RNA-seq, microarrays |
Pronóstico de mortalidad, subclasificación fenotípica |
|
Tisular o celular |
TGF-β, α-SMA, CD14+, colágeno tipo I |
Biopsia pulmonar, LBA / inmunohistoquímica, citometría de flujo |
Confirmación diagnóstica, estudios de mecanismos patogénicos |
Abreviaturas: FPI: fibrosis pulmonar idiopática; CVF: capacidad vital forzada; DLCO: capacidad de difusión de monóxido de carbono; LBA: lavado broncoalveolar; TCAR: tomografía computarizada de alta resolución; SNP: polimorfismo de nucleótido único; SGN: secuenciación de nueva generación; CD14+: marcador de superficie de monocitos; α-SMA: alfa actina de músculo liso; TGF-β: factor de crecimiento transformante beta; SP-A/SP-D: proteínas surfactantes A y D; MMP-7: metaloproteinasa de matriz 7; MUC5B: Mucina 5B; TERT: Transcriptasa inversa de la telomerasa; TERC: Telomerasa RNA; NIU: neumonía intersticial usual.
Tabla 2 .
Clasificación de biomarcadores en la FPI.
|
Clasificación |
Biomarcadores implicados |
|---|---|
|
Biomarcadores expresados diferencialmente en la FPI frente a sujetos sanos |
KL-6, SP-A, SP-D, MMP-1, MMP-7, MMP-28, POSTN, OPN, YKL-40, RAGE, SAA |
|
Biomarcadores para el diagnóstico diferencial entre FPI y otras EPID |
MMP-28, POSTN, OPN, MMP-1, MMP-7 |
|
Biomarcadores que diferencian la exacerbación aguda de la FPI, de la enfermedad estable |
KL-6, SP-A, SP-D, S100A9, α-defensina, CXCL8, CXCL13, CCL2, IL-6, LTBP2, HMGB1 |
|
Biomarcadores que predicen la progresión de la enfermedad |
KL-6, SP-A, SP-D, RAGE, MMP-7, POSTN, YKL-40, monocitos |
|
Biomarcadores relacionados con la gravedad de la enfermedad |
KL-6, RAGE, SAA, CEA, MMP-7, YKL-40, VEGF, monocitos |
|
Biomarcadores relacionados con el tratamiento |
IGFBP-2, Gal-3, SP-D |
Abreviaturas: FPI: fibrosis pulmonar idiopática; EPID: enfermedad pulmonar intersticial difusa; MEC: matriz extracelular; CEA: células epiteliales alveolares; DLCO: capacidad de difusión de monóxido de carbono; CVF: capacidad vital forzada; LBA: lavado broncoalveolar; KL-6: Krebs von den Lungen-6; SP-A / SP-D: proteínas del surfactante A / D; MMP: metaloproteinasas de la matriz; POSTN: periostina; OPN: osteopontina; SAA: proteína amiloide sérica A; RAGE: receptor de productos finales de glicación avanzada; CCL2 (MCP-1): proteína quimioatrayente de monocitos-1; CXCL8: interleucina-8; HMGB1: proteína de unión a cajas de alta movilidad 1; VEGF: factor de crecimiento endotelial vascular; IGFBP-2: proteína de unión al factor de crecimiento similar a la insulina tipo 2; LTBP2: proteína de unión al factor de crecimiento transformante beta 2; YKL-40: quitinasa 3 similar a la proteína 1.
El biomarcador ideal debe ser específico, sensible, predictivo, rápido y económico, estable in vivo e in vitro, no invasivo, y que tenga suficiente relevancia preclínica y clínica como para modificar las decisiones relativas al proceso patológico en que se aplica.27 La mayoría de los biomarcadores mencionados no cumplen con todos los criterios y no son accesibles de forma rutinaria en muchas regiones del mundo, por lo cual los monocitos podrían convertirse en una opción asequible.
El rol de los monocitos en el desarrollo y progresión de la fibrosis pulmonar idiopática
Los monocitos inflamatorios y los macrófagos tisulares residentes desempeñan una función reguladora fundamental en los procesos de reparación, regeneración y fibrosis tisular. Su participación en la patogénesis de la fibrosis pulmonar idiopática está bien establecida. Esta implicación se ve reforzada por la correlación entre elevados recuentos monocitarios y mayor mortalidad en otras patologías fibróticas, incluyendo esclerosis sistémica, miocardiopatía hipertrófica y mielofibrosis,8 lo que sugiere un posible mecanismo patogénico común.
Tras una lesión tisular, los monocitos y macrófagos experimentan cambios fenotípicos y funcionales profundos que les permiten cumplir funciones esenciales durante las fases de inicio, mantenimiento y resolución de la reparación tisular. Las disfunciones en la actividad de los monocitos pueden conllevar a una reparación inadecuada, caracterizada por la liberación desregulada de mediadores inflamatorios y factores de crecimiento, la generación insuficiente de poblaciones de monocitos con perfil antiinflamatorio o la alteración en la comunicación con células epiteliales, endoteliales, fibroblastos y células madre o progenitoras. Estas alteraciones favorecen un microambiente persistente de lesión, que finalmente puede derivar en el desarrollo de fibrosis patológica.28
Diferenciación y activación de los monocitos en la FPI
En respuesta a la lesión de las células epiteliales alveolares, se liberan citocinas que reclutan células inflamatorias, incluyendo monocitos. Las vías de señalización de quimiocinas, como CXCL12/SDF1-CXCR4, CCL21-CCR7 y CCL2-CCR2, desempeñan un papel importante en el reclutamiento de fibrocitos (progenitores de monocitos) y células progenitoras mesenquimales a los pulmones. En particular, CCL2 y CCL3 participan en la migración de monocitos/macrófagos hacia los pulmones. Se ha demostrado que el bloqueo de estas moléculas puede tener un efecto antifibrótico.3 (Figura 1)
Figura 1.
Monocitos-macrófagos y su papel en la fibrosis del pulmón.
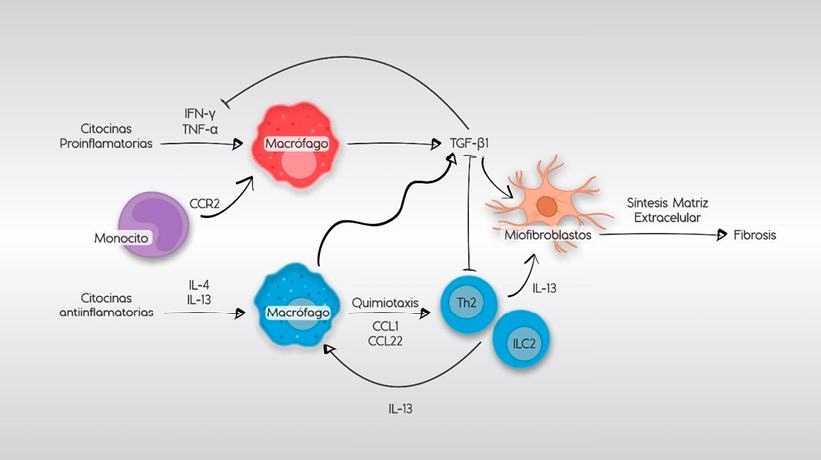
Creada con BioRender.com
Los monocitos activos por un factor desencadenante liberan citocinas proinflamatorias que activan al macrófago, pero concomitantemente citocinas antiinflamatorias también se activan como mecanismo reparador. Sin embargo, ante un estímulo persistente, también contribuyen a la activación de miofibroblastos, que con la síntesis de matriz extracelular contribuyen a la fibrosis.
Una vez en el pulmón, los monocitos circulantes pueden diferenciarse en distintos tipos celulares:
- Macrófagos derivados de monocitos (Mo-AMs): Estos macrófagos alveolares derivados de monocitos surgen lentamente en un proceso que implica la represión de genes específicos de monocitos y la activación de genes característicos de macrófagos alveolares. Datos transcriptómicos indican que esta diferenciación está regulada por la reprogramación epigenética impulsada por el microambiente pulmonar. En modelos murinos, la protección contra la fibrosis inducida por bleomicina se asocia con una reducción de los Mo-AMs.29
- Fibrocitos: Además, los fibrocitos (progenitores de monocitos) pueden diferenciarse en fibroblastos, proceso observado in vitro con MEC de pacientes con FPI. Factores como TGF-β, endotelina, CTGF, IL-3, IL-4, factores de respuesta del suero y algunos microRNAs favorecen esta diferenciación. El proteoglicano lumican también participa en este proceso a través de una vía dependiente de integrinas.30
Aunque la polarización clásica M1/M2 ((M1 macrófago proinflamatorio) y (M2 macrófago reparador/profibrótico)) es característica de macrófagos, los monocitos circulantes también tienen un papel activo en la patogénesis de la FPI, contribuyendo a un microambiente profibrótico o antifibrótico mediante la secreción de mediadores y su interacción con otras células:31
- Los monocitos producen MMPs con efectos duales: MMP8, con efectos profibróticos, y MMP19, con propiedades antifibróticas, relacionadas con la cicatrización y migración celular.32
- La actividad de monocitos y macrófagos puede ser modulada por proteínas como la pentraxina-2 (PTX-2), la cual ejerce un efecto regulador sobre estos, especialmente al reducir la actividad profibrótica de los macrófagos tipo M2.33
- Los fibrocitos que se diferencian en fibroblastos contribuyen significativamente a la secreción excesiva de MEC, y desempeñan así un papel central en la progresión de la fibrosis pulmonar.29
Rol de los monocitos en la inflamación crónica
Los monocitos son activamente reclutados en el parénquima pulmonar afectado por la FPI. Este proceso es esencial en la progresión de la FPI, ya que contribuye a la acumulación de fibroblastos y a la deposición de MEC, y genera así un entorno profibrótico.34 Estos monocitos muestran un fenotipo inmunológico alterado caracterizado por una mayor expresión de CD64 (FcγR1), la cual se ha correlacionado con el grado de fibrosis pulmonar, sugiriendo que estos están “preparados” para participar activamente en la respuesta inflamatoria y fibrogénica.35 Además, los macrófagos alternativos (M2) derivados de monocitos desempeñan un papel clave en la perpetuación de la fibrosis, siendo los monocitos Ly6Chi especialmente relevantes en la facilitación de un microambiente profibrótico.36 Estudios también han demostrado que los sueros de pacientes con FPI pueden estimular la angiogénesis mediada por monocitos, lo que implica un rol adicional en la formación de nuevos vasos sanguíneos en el contexto de la inflamación crónica.37 Asimismo, tanto los monocitos como los macrófagos derivados de estos están implicados en la producción de factores de crecimiento, como el heparin-binding epidermal growth factor-like growth factor (HB-EGF), que se asocia con la activación de macrófagos con un perfil profibrótico.38 La complejidad de la respuesta inflamatoria y la plasticidad funcional de estos monocitos y macrófagos explican, en parte, la limitada eficacia de las terapias antiinflamatorias convencionales y subrayan la necesidad de abordar vías patogénicas más específicas, como lo ejemplifican los estudios actuales con agentes que regulan la actividad de estas células, tales como PRM-151.39
Interacción de los monocitos con otros mediadores fibróticos
Las interacciones entre los monocitos y otros mediadores fibróticos en la FPI son complejas y multifacéticas, e incluyen procesos como la diferenciación celular, la señalización paracrina y el reclutamiento celular. Estas interacciones son fundamentales para la progresión de la enfermedad y representan potenciales dianas terapéuticas. Los monocitos reclutados en el microambiente pulmonar dañado pueden diferenciarse en macrófagos intersticiales (Mo-IMs) con un fenotipo profibrótico. Estos macrófagos expresan genes asociados a la reparación y la remodelación de la MEC, contribuyendo activamente a la patogénesis de la enfermedad.29 Los Mo-IMs interactúan directamente con fibroblastos mediante vías de señalización clave como TGF-β, SPP1 y PDGF (factor de crecimiento derivado de plaquetas), lo que promueve así la progresión de la fibrosis. Además, los monocitos derivados de pacientes con FPI muestran una hipersensibilidad a estímulos como CHI3L1, lo que incrementa la expresión de marcadores profibróticos, como CD206, y sugiere su posible utilidad como biomarcador predictivo de respuesta a intervenciones terapéuticas.40
El reclutamiento de monocitos Ly6Chi, caracterizados por un fenotipo proinflamatorio, se ve estimulado por la exposición a partículas finas (PM2.5) y contribuye a la exacerbación y progresión de la fibrosis. Por otro lado, los fibrocitos, que derivan de monocitos, juegan un papel adicional al producir factores de crecimiento como PDGF, que estimulan la proliferación de fibroblastos en los pulmones afectados.41
La quimiotaxis y la infiltración de monocitos/macrófagos son procesos mediados por quimiocinas clave como lo es CCL2, CCL3 y CCL7 que están particularmente involucradas en la migración y activación de estos leucocitos, y se encuentran elevados en pacientes con FPI. Aunque el antagonismo de estas quimiocinas se ha postulado como estrategia antifibrótica, los resultados clínicos han sido contradictorios.8 Asimismo, las vías CXCL12/SDF1-CXCR4 y CCL21-CCR7 regulan el reclutamiento de fibrocitos circulantes y células progenitoras mesenquimales, subrayando la complejidad de la respuesta inflamatoria y fibrogénica en la FPI.42
Los macrófagos derivados de monocitos son una fuente relevante de factores de crecimiento y citocinas profibróticas, incluyendo TGF-β, un mediador central en la fibrosis, cuya producción está fuertemente asociada con la actividad de los macrófagos M2. Otras moléculas implicadas son IL-8, que incrementa la fibrogenicidad de células progenitoras mesenquimales y estimula su proliferación y activación, y factores de crecimiento como FGF, PDGFα, IGF1 y VEGF, que promueven la angiogénesis y consolidan un microambiente profibrótico.8 En este sentido, la vía de señalización de M-CSF/M-CSFR ha emergido como un regulador clave para mantener nichos fibróticos espacialmente restringidos y representa una potencial diana terapéutica en el manejo de los Mo-AMs.43
Por otra parte, el factor de crecimiento HB-EGF, derivado de células mieloides y epiteliales, se ha vinculado con la fibrosis pulmonar y podría influir en la migración de monocitos mediante la inducción de CCL2.38 Estos monocitos y macrófagos participan activamente en la remodelación de la MEC, siendo una fuente importante de MMPs como MMP1, MMP3, MMP8, MMP19 y MMP28. Estas MMPs, junto con sus inhibidores (TIMPs), regulan finamente la degradación de la MEC y modulan la actividad fibrogénica.44 Algunas, como MMP8 y MMP28, favorecen la polarización de macrófagos hacia un fenotipo M2, mientras que otras, como MMP19 y MMP14, tienen efectos antifibróticos y contribuyen a la activación de TGF-β y la colagenólisis.45 (Figura 2)
Figura 2.
Citoquinas y quimiocinas que intervienen en la activación de monocitos.
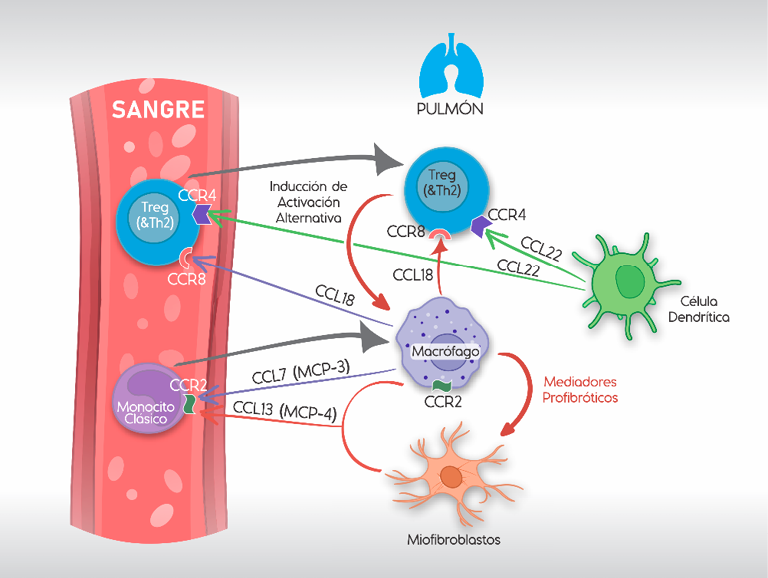
Creada con BioRender.com
Para la activación y acumulación de monocitos y concomitantemente de Tregs (células T reguladoras), están implicados factores estimulantes de colonias (M-CSF, G-CSF, GM-CSF) y proteínas quimioatrayentes de monocitos (MCPs:CCL2, CCL7, CCL8, CCL13) las cuales se unen al ligando CCR2, el principal receptor quimioatrayente de monocitos. Para Tregs, se destacan los receptores CCR4 y CCR8, para ligandos de quimiocina CCL18 y CCL22. La expresión de CCL22 es dada principalmente por las células dendríticas del pulmón, los macrófagos expresan predominantemente CCL18 y CCL7. CCL13 es expresado por los mismos miofibroblastos y macrófagos.
Finalmente, los monocitos no solo activan miofibroblastos mediante la secreción de mediadores como TGF-β e IL-8, sino que también modulan directamente la función de los fibroblastos, lo que promueve su activación y proliferación. En conjunto, estas complejas interacciones entre monocitos, macrófagos y otros mediadores fibróticos resaltan la importancia de estas células en la fisiopatología de la FPI.46
Monocitos como biomarcador en FPI
Evidencia del recuento de monocitos como biomarcador en FPI
La FPI tiene un curso progresivo e impredecible, por lo que los biomarcadores permitirían identificar pacientes con riesgo acelerado de progresión, exacerbaciones agudas y muerte. En ese marco, el recuento absoluto de monocitos en sangre periférica ha emergido como un biomarcador pronóstico atractivo por una serie de razones clínicas y operativas que lo distinguen de otros marcadores moleculares o en investigación.47
Accesibilidad y bajo costo: el recuento de monocitos forma parte del hemograma completo, una prueba universalmente disponible en la práctica clínica y de bajo costo, sin necesidad de plataformas moleculares especializadas ni procesamiento adicional.48
Reproducibilidad y estandarización: a diferencia de biomarcadores séricos menos estandarizados (KL-6, SP-D, MMPs), el recuento absoluto de monocitos se mide de forma automatizada con parámetros bien definidos, lo que favorece su comparabilidad interlaboratorio.31
Estabilidad relativa en el tiempo: a diferencia de otras variables clínicas que pueden fluctuar con el tratamiento o las exacerbaciones, el recuento de monocitos ha demostrado mantenerse relativamente estable a lo largo del tiempo, lo que lo posiciona como un marcador basal útil para estratificación de riesgo.52
Valor predictivo independiente: múltiples estudios han evidenciado que su capacidad pronóstica es independiente de parámetros clásicos como la CVF, la DLCO o el índice GAP, por lo que aporta un valor clínico adicional.47,49
Aplicabilidad transversal: más allá de la FPI, el recuento de monocitos también ha demostrado utilidad pronóstica en otras enfermedades pulmonares fibrosantes y enfermedades sistémicas con componente intersticial, como la esclerosis sistémica, lo que sugiere un rol transversal en la fisiopatología de la fibrosis.50
Relación entre monocitos y desenlaces clínicos
Riesgo de mortalidad
Un recuento elevado de monocitos se asocia con un mayor riesgo de mortalidad por todas las causas. Un metaanálisis encontró que los HR ajustados para la mortalidad por recuentos de monocitos ≥ 950 células/µL fue de 1,93 (IC 95% 1,24–3,01).7 Otro análisis agrupado de ensayos clínicos de fase III (ASCEND, CAPACITY, INSPIRE) mostró que los pacientes con un recuento de monocitos de 600 a <950 células/µL o ≥ 950 células/µL tuvieron un riesgo significativamente mayor a 1 año de mortalidad, en comparación con aquellos con recuentos <600 células/µL. Incluso, se incluye una modificación al modelo de GAP clásico que incluye edad, sexo y función pulmonar, y asocia el conteo de monocitos, glóbulos rojos y neutrófilos. Esta modificación proporciona estadísticas nominales más altas para la progresión de la FPI, la mortalidad por todas las causas, la hospitalización por todas las causas y la hospitalización relacionada con problemas respiratorios, lo que demuestra una mejora sobre los modelos previos.50 El umbral de ≥ 600 células/µL ha sido sugerido como un punto de corte robusto y reproducible asociado a un mayor riesgo de mortalidad basado en este trabajo.
En un estudio retrospectivo realizado por Coelho et al., en una cohorte de 96 pacientes seguidos durante al menos seis meses, se observó que aquellos con un recuento basal de monocitos ≥950 células/µL presentaron una supervivencia significativamente menor en comparación con quienes tenían valores inferiores (14,3 vs. 28,9 meses; p = 0,004). Aunque el tratamiento antifibrótico indujo una reducción significativa del recuento monocitario a los seis meses, este efecto no se mantuvo en seguimientos más prolongados.51
En un análisis multicéntrico y multietapa, utilizando datos transcriptómicos de 120 muestras de células mononucleares en sangre periférica, los autores identificaron que un porcentaje elevado de monocitos CD14+ se asociaba con menor supervivencia libre de trasplante. Estos hallazgos fueron validados en dos cohortes independientes (COMET y Yale), donde recuentos elevados de monocitos se correlacionaron con mayor riesgo de progresión y peor pronóstico clínico. Además, en un análisis de registros electrónicos de salud de más de 45.000 pacientes con FPI u otras enfermedades fibróticas, un recuento absoluto de monocitos ≥950 células/µL se asoció de manera consistente con mayor mortalidad, incluso tras ajustar por función pulmonar y el índice GAP.52
Progresión de la enfermedad
Con relación al conteo de monocitos y el riesgo de progresión o exacerbación de la enfermedad, se destaca el estudio retrospectivo de Karampitsakos y et al., donde se evaluó el recuento de monocitos y la amplitud de distribución eritrocitaria (ADE), en pacientes con FPI sin tratamiento previo. Analizando dos cohortes independientes (n=489), los autores demostraron que un recuento de monocitos ≥600 células/µL se asoció con una menor función pulmonar basal (CVF y DLCO) y una mayor mortalidad por todas las causas, con HR de 2,05 (IC 95%: 1,19–3,53; p = 0,01). Estos hallazgos fueron validados en una segunda cohorte con resultados comparables. La ADE elevada (≥14,1%) también mostró una asociación significativa con peor función pulmonar.49 En un estudio retrospectivo, Achaiah et al. evaluaron a pacientes con patrón tomográfico indeterminado para NIU (neumonía intersticial usual), considerado un posible estadio temprano de FPI. De los pacientes con NIU indeterminada evaluados, 53% progresó a FPI en un promedio de 4 años. El análisis mostró que un recuento de monocitos en sangre periférica superior a 900 células/µL al momento del diagnóstico se asociaba de forma significativa con dicha progresión (HR: 23; IC 95%: 1,6–340; p = 0,03), al igual que niveles elevados de neutrófilos.57 En un estudio de pacientes FPI, se evaluó el valor pronóstico de parámetros sanguíneos de rutina y biomarcadores bioquímicos relacionados con inflamación, con el objetivo de desarrollar un modelo predictivo robusto. A través de un análisis multivariado en 377 pacientes, se demostró que tanto el recuento absoluto de monocitos como la razón monocito/glóbulo rojo (MGR) se asociaron significativamente con un mayor riesgo de mortalidad. A partir de estos hallazgos, los autores desarrollaron y validaron un modelo pronóstico denominado índice y estadio CPB (clinical-physiological-biomarker), que integra variables clínicas (edad, sexo), funcionales (CVF y DLCO porcentuales) y biomarcadores inflamatorios, destacando el valor del conteo de monocitos.47
Bernardinello et al. analizaron tres cohortes distintas de pacientes con FPI: recién diagnosticados, en estadio terminal y con FPI asociada a cáncer de pulmón; evaluaron el valor pronóstico del recuento absoluto de monocitos y de la relación linfocito/monocito (RLM). En los pacientes recién diagnosticados, un recuento elevado de monocitos mostró una correlación negativa con la CVF al momento del diagnóstico (r = -0,27; p = 0,01) y se asoció de manera independiente con un mayor riesgo de deterioro funcional (OR: 1,004 IC 95% 1,00–1,01; p = 0,03). Asimismo, un RLM <4,18 se asoció con una peor supervivencia global (HR: 6,88; IC 95% 2,55–18,5; p = 0,027), con un área bajo la curva de 0,67 para la predicción de desenlace. De forma adicional, el RLM fue significativamente más bajo en pacientes con FPI y cáncer de pulmón, en comparación con aquellos recién diagnosticados o en fase terminal de la enfermedad.53
Riesgo de exacerbación
Con el propósito de evaluar la relación entre los monocitos y el riesgo de exacerbación, se destaca el estudio de cohorte retrospectivo de Kawamura et al. en el que se analizó el impacto del recuento absoluto de monocitos en la aparición de exacerbaciones agudas (EA) en pacientes con enfermedades pulmonares intersticiales fibrosantes, incluidos aquellos con FPI tratados con agentes antifibróticos. En una muestra de 122 pacientes sin antecedentes de EA, se observó que aquellos con mayor recuento de monocitos al inicio del tratamiento presentaron un riesgo significativamente más alto de desarrollar una EA, así como un tiempo más corto hasta su aparición. Esta asociación se mantuvo como un factor independiente incluso tras ajustar por la gravedad de la enfermedad. Con base en estos resultados, los autores propusieron un sistema de puntuación simple que combina el recuento de monocitos (umbral: 380 células/µL) con el puntaje GAP, como posible herramienta predictiva de exacerbación.54
Respuesta terapéutica y perfil de eventos adversos
El conteo de monocitos se ha estudiado para evaluar la tolerabilidad a la terapia antifibrótica. En un estudio de cohorte retrospectivo que evaluó a 111 pacientes con enfermedad pulmonar intersticial incluyendo FPI tratados con nintedanib, se identificó que un recuento de monocitos superior a 454 células/µL se asoció significativamente con un mayor riesgo de fracaso del tratamiento, definido como reducción de dosis, retiro temporal o suspensión definitiva del fármaco, incluso bajo manejo sintomático adecuado. Este hallazgo posiciona al recuento de monocitos como un factor de riesgo comparable al área de superficie corporal en la tolerancia al tratamiento. En cuanto a la eficacia, no se observaron diferencias en la frecuencia de exacerbaciones agudas ni en el deterioro funcional CVF entre pacientes que iniciaron con dosis estándar (300 mg) frente a dosis reducidas (200 mg). Estos resultados sugieren que en pacientes con monocitos elevados o contextura baja, podría considerarse una dosis inicial reducida de nintedanib para minimizar eventos adversos sin comprometer su eficacia terapéutica.55 (Tabla 3)
Tabla 3.
Estudios relevantes sobre monocitos como biomarcador en la FPI.
|
Autor y año |
País del estudio |
Edad, años |
Hombre % |
CVF % predicho, basal |
DLco % predicho, basal |
Conteo monocitos |
Momento de la medida monocitos |
Resultados relevantes |
|---|---|---|---|---|---|---|---|---|
|
Scott et al.52 2019 |
Estados unidos |
NA |
NA |
NA |
NA |
⩾950 células/µL |
En los 30 días al diagnóstico |
Sobrevida, tiempo libre de trasplante |
|
Teoh et al.31 2020 |
Australia |
69,9±8,3 |
71 |
80,3±22 |
48,2±16,8 |
⩾950 células/µL |
Al inicio del diagnóstico |
Sobrevida |
|
Karampitsakos et al.49 2021 |
Multinacional |
NA |
NA |
NA |
NA |
⩾950 células/µL (agrupado) ⩾600 células/µL (inicio y validación) |
Previo a inicio de antifibrótico |
Mortalidad por todas las causas, progresión de la enfermedad a 1 año, por deterioro funcional |
|
Kreuter et al.48 2021 |
Estados Unidos |
NA |
NA |
NA |
NA |
600-800 células/µL ⩾950 células/µL |
Al inicio del diagnóstico |
Todas las causas de mortalidad a 1 año |
|
Achaiah et al.57 2022 |
Reino Unido |
74,8±6,9 |
79 |
85,5 (69,9–98,0) |
61,9 (50,9–71,0) |
>900 células/µL |
En los primeros 4 meses de la presentación clínica |
Caída de la CVF >10% por año, mortalidad por todas las causas |
|
Zhang et al.47 2022 |
China |
64,5±9,46 |
82,69 |
77,39±20,31 |
40,36±18,71 |
>670 células/µL |
Después de la admisión al hospital |
Sobrevida |
|
Bernardinello et al.53 2022 |
Italia |
70 (53–81) |
83 |
80 (50–125) |
57 (30–106) |
RLM medición continua |
Al diagnóstico y en el seguimiento de primer año de terapia antifibrótica |
Caída ≥ 5% de la CVF en el seguimiento a 1 año |
|
Tsuneyoshi S et al.55 2023 |
Japón |
70 (66–75) |
72,1 |
70,4 (57,1–87,4) |
56,3 (44,8–73,7) |
451 (352– 600) células/µL |
Al inicio y seguimiento del tratamiento con nintedanib |
Mayor efectos secundarios con el tratamiento |
|
Kawamura et al.54 2020 |
Japón |
68(65,73) |
73 |
74,3 |
58,6 |
≥380 células/µL |
Al inicio del tratamiento |
Riesgo de exacerbación |
Abreviaturas: NA: no aplica; CVF: capacidad vital forzada; RLM: relación linfocito monocito; DCLO: capacidad de difusión de monóxido de carbono.
Limitaciones y desafíos en su implementación clínica
Uno de los principales desafíos es la necesidad de una validación más extensa y rigurosa. A pesar de los estudios que muestran una asociación entre el recuento elevado de monocitos y un mayor riesgo de mortalidad y progresión de la enfermedad, se requieren estudios prospectivos, a gran escala, multicéntricos y en entornos del mundo real para confirmar estos hallazgos, evaluar su papel pronóstico a largo plazo y determinar su relación con la respuesta a los tratamientos antifibróticos.49 Las asociaciones específicas, como la vinculación con las exacerbaciones agudas o los eventos adversos del tratamiento, también necesitan ser validadas en cohortes más amplias.3
Muchos estudios son de naturaleza retrospectiva, lo que introduce posibles sesgos y factores de confusión.3 La inclusión de poblaciones heterogéneas (con diferentes tipos de enfermedad pulmonar intersticial, gravedad de la enfermedad o estado del tratamiento) dificulta la comparación directa de los resultados.51 El uso de tamaños muestrales moderados o un número limitado de eventos (como exacerbaciones agudas) en algunos estudios puede reducir la potencia estadística y limitar la posibilidad de análisis multivariantes completos. Además, existen limitaciones en los datos recopilados en algunos estudios, como la falta de información radiológica, la causa específica de la muerte, los niveles de Pao2 (presión arterial de oxígeno) o el uso detallado de corticosteroides u otros tratamientos concomitantes que pueden influir en el recuento de monocitos.49
Aunque se ha implicado a los monocitos y macrófagos en la patogénesis de la FPI, los mecanismos precisos que subyacen a su asociación con los resultados clínicos aún necesitan ser desentrañados.8 Comprender el papel de subpoblaciones específicas de monocitos/macrófagos y sus fenotipos (como macrófagos derivados de monocitos frente a macrófagos residentes, o fenotipos pro-fibróticos específicos) es importante y añade una capa de complejidad más allá del simple recuento total. Para abordar esta pregunta es necesario ampliar en técnicas avanzadas como secuenciación de ARN de células individuales, citometría de alto rendimiento y técnicas de trazado de linaje.56
Conclusión
Los estudios más representativos coinciden en que el recuento de monocitos en sangre periférica constituye un biomarcador pronóstico emergente en la FPI. Diversos trabajos multicéntricos y de cohortes amplias han demostrado que valores elevados de monocitos se asocian con mayor mortalidad, mayor declive funcional y riesgo aumentado de exacerbaciones agudas. Su integración dentro de índices clínicos y fisiológicos, como el GAP o el CPB, ha permitido mejorar la estratificación pronóstica, con especial relevancia en pacientes que reciben tratamiento antifibrótico continuo.47,50 La facilidad para cuantificar los monocitos mediante hemograma completo, su bajo costo y amplia disponibilidad lo posicionan como un candidato atractivo para complementar los modelos clínicos tradicionales.
No obstante, persisten importantes limitaciones metodológicas que dificultan su implementación rutinaria. La falta de estandarización en los puntos de corte, la heterogeneidad de las poblaciones estudiadas y la ausencia de validación prospectiva en contextos diversos limitan la reproducibilidad de los hallazgos. A ello se suma la influencia de factores confusores como comorbilidades, uso de corticosteroides o infecciones intercurrentes que pueden alterar el valor del biomarcador. Además, el vínculo fisiopatológico entre la monocitosis periférica y la progresión fibrótica no se comprende completamente; la evidencia actual sugiere un papel activo de los monocitos y macrófagos intersticiales en la activación de vías profibróticas mediadas por TGF-β, IL-6 y CCL2, especialmente en condiciones de hipoxia crónica.8,42
Desde una perspectiva crítica, el valor del recuento de monocitos debe interpretarse en el contexto de su integración con otros biomarcadores celulares y moleculares, más que como un parámetro independiente. La combinación de parámetros hematológicos (como el índice linfocito/monocito o el MGR) con marcadores séricos de remodelación tisular (MMP-7, KL-6, periostina) podría ofrecer un perfil pronóstico más robusto y reproducible.4,42,53 En el futuro, los estudios multicéntricos prospectivos deberán definir umbrales uniformes y validar su capacidad predictiva dentro de estrategias de medicina personalizada.
En síntesis, aunque el recuento de monocitos es un marcador prometedor, su adopción clínica debe basarse en evidencia armonizada y mecanismos biológicos claramente definidos. Su validación definitiva podría traducirse en una mejora sustancial del abordaje individualizado, la predicción de exacerbaciones y la toma de decisiones terapéuticas en la FPI.
Financiamiento: el autor declara que el trabajo no tuvo financiamiento.
Conflictos de interés: el autor declara que no tiene conflictos de intereses relacionados con el tema de esta publicación.
Contribuciones de los autores: no corresponde.
Los Editores en Jefe, Dres. Carlos Luna y Francisco Arancibia, realizaron el seguimiento del proceso de revisión y aprobaron este artículo.
Referencias
1. Maher TM, Bendstrup E, Dron L, Langley J, Smith G, Khalid JM et al. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res 2021;22(1):197. https://doi.org/10.1186/s12931-021-01791-z
2. Zhu W, Liu C, Tan C, Zhang J. Predictive biomarkers of disease progression in idiopathic pulmonary fibrosis. Heliyon 2023;10(1):e23543. https://doi.org/10.1016/j.heliyon.2023.e23543
3. Jee AS, Sahhar J, Youssef P, Bleasel J, Adelstein S, Nguyen M et al. Review: Serum biomarkers in idiopathic pulmonary fibrosis and systemic sclerosis associated interstitial lung disease - frontiers and horizons. Pharmacol Ther 2019;202:40-52. https://doi.org/10.1016/j.pharmthera.2019.05.014
4. Wang Q, Xie Z, Wan N, Yang L, Jin Z, Jin F et al. Potential biomarkers for diagnosis and disease evaluation of idiopathic pulmonary fibrosis. Chin Med J (Engl) 2023;136(11):1278-90. https://doi.org/10.1097/CM9.0000000000002171
5. Guiot J, Moermans C, Henket M, Corhay JL, Louis R. Blood biomarkers in idiopathic pulmonary fibrosis. Lung 2017;195(3):273-80. https://doi.org/10.1007/s00408-017-9993-5
6. Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183(4):431-40. https://doi.org/10.1164/rccm.201006-0894CI
7. Min B, Grant-Orser A, Johannson KA. Peripheral blood monocyte count and outcomes in patients with interstitial lung disease: a systematic review and meta-analysis. Eur Respir Rev 2023;32(169):230072. https://doi.org/10.1183/16000617.0072-2023
8. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 2017;214(8):2387-404. https://doi.org/10.1084/jem.20162152
9. Phan THG, Paliogiannis P, Nasrallah GK, Giordo R, Eid AH, Fois AG et al. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell Mol Life Sci 2021;78(5):2031-57. https://doi.org/10.1007/s00018-020-03665-0
10. She YX, Yu QY, Tang XX. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov 2021;7(1):52. https://doi.org/10.1038/s41420-021-00426-2
11. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 2003;112(12):1776-84. https://doi.org/10.1172/JCI20530
12. Tsoutsou PG, Gourgoulianis KI, Petinaki E, Germenis A, Tsoutsou AG, Mpaka M et al. Cytokine levels in the sera of patients with idiopathic pulmonary fibrosis. Respir Med 2006;100(5):938-45. https://doi.org/10.1016/j.rmed.2005.08.010
13. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002;3(5):349-63. https://doi.org/10.1038/nrm809
14. Todd NW, Scheraga RG, Galvin JR, Iacono AT, Britt EJ, Luzina IG et al. Lymphocyte aggregates persist and accumulate in the lungs of patients with idiopathic pulmonary fibrosis. J Inflamm Res 2013;6:63-70. https://doi.org/10.2147/JIR.S44170
15. Moua T, Lee AS, Ryu JH. Comparing effectiveness of prognostic tests in idiopathic pulmonary fibrosis. Expert Rev Respir Med 2019;13(10):993-1004. https://doi.org/10.1080/17476348.2019.1661357
16. White ES, Xia M, Murray S, Dyal R, Flaherty CM, Flaherty KR et al. Plasma surfactant protein-D, matrix metalloproteinase-7, and osteopontin index distinguishes idiopathic pulmonary fibrosis from other idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2016;194(10):1242-51. https://doi.org/10.1164/rccm.201505-0862OC
17. Xu X, Chen H, Zhu X, Ma Y, Liu Q, Xue Y et al. S100A9 promotes human lung fibroblast cells activation through receptor for advanced glycation end-product-mediated extracellular-regulated kinase 1/2, mitogen-activated protein-kinase and nuclear factor-κB-dependent pathways. Clin Exp Immunol 2013;173(3):523-35. https://doi.org/10.1111/cei.12118
18. Papiris SA, Tomos IP, Karakatsani A, Spathis A, Korbila I, Analitis A et al. High levels of IL-6 and IL-8 characterize early-on idiopathic pulmonary fibrosis acute exacerbations. Cytokine 2018;102:168-72. https://doi.org/10.1016/j.cyto.2017.08.019
19. Fathimath Muneesa M, Shaikh SB, Jeena TM, Bhandary YP. Inflammatory mediators in various molecular pathways involved in the development of pulmonary fibrosis. Int Immunopharmacol. 2021;96:107608. https://doi.org/10.1016/j.intimp.2021.107608
20. Kayıkçı A, Alatas F, Alatas IO, Yıldırım H, Ozen H. The role of biomarkers in the diagnosis and treatment follow-up of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2024;41(2):e2024015. https://doi.org/10.36141/svdld.v41i2.15454
21. Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest 2013;143(5):1422-9. https://doi.org/10.1378/chest.12-1134
22. Hamai K, Iwamoto H, Ishikawa N, Horimasu Y, Masuda T, Miyamoto S et al. Comparative study of circulating MMP-7, CCL18, KL-6, SP-A, and SP-D as disease markers of idiopathic pulmonary fibrosis. Dis Markers 2016;2016:4759040. https://doi.org/10.1155/2016/4759040
23. Kalafatis D, Löfdahl A, Näsman P, Dellgren G, Wheelock ÅM, Elowsson-Rendin L et al. Distal lung microenvironment triggers release of mediators recognized as potential systemic biomarkers for idiopathic pulmonary fibrosis. Int J Mol Sci 2021;22(24):13421. https://doi.org/10.3390/ijms222413421
24. Guiot J, Bondue B, Henket M, Corhay JL, Louis R. Raised serum levels of IGFBP-1 and IGFBP-2 in idiopathic pulmonary fibrosis. BMC Pulm Med 2016;16(1):86. https://doi.org/10.1186/s12890-016-0243-4
25. Jenkins RG, Cottin V, Nishioka Y, Noth I, White ES, Ittrich C et al. Effects of nintedanib on circulating biomarkers of idiopathic pulmonary fibrosis. ERJ Open Res 2024;10(6):00558-2023. https://doi.org/10.1183/23120541.00558-2023
26. Koga Y, Motegi M, Ono A, Hachisu Y, Utsugi M, Sunaga N et al. Serum galectin-3 as a biomarker of progression of idiopathic pulmonary fibrosis treated with nintedanib. Respir Investig 2025;63(3):394-8. https://doi.org/10.1016/j.resinv.2024.12.005
27. Martín-Ventura JL, Blanco-Colio LM, Tuñón J, Muñoz-García B, Madrigal-Matute J, Moreno JA et al. Biomarkers in cardiovascular medicine. Rev Esp Cardiol 2009;62(6):677-88. https://doi.org/10.1016/s1885-5857(09)72232-7
28. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016;44(3):450-62. https://doi.org/10.1016/j.immuni.2016.02.015
29. Wang S, Li J, Wu C, Lei Z, Wang T, Huang X et al. Single-cell RNA sequencing reveals monocyte-derived interstitial macrophages with a pro-fibrotic phenotype in bleomycin-induced pulmonary fibrosis. Int J Mol Sci 2024;25(21):11669. https://doi.org/10.3390/ijms252111669
30. Ghanem M, Justet A, Jaillet M, Vasarmidi E, Boghanim T, Hachem M et al. Identification of FGFR4 as a regulator of myofibroblast differentiation in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2024;327(6):L818-30. https://doi.org/10.1152/ajplung.00184.2023
31. Teoh AKY, Jo HE, Chambers DC, Symons K, Walters EH, Goh NS et al. Blood monocyte counts as a potential prognostic marker for idiopathic pulmonary fibrosis: analysis from the Australian IPF registry. Eur Respir J 2020;55(4):1901855. https://doi.org/10.1183/13993003.01855-2019
32. Craig VJ, Zhang L, Hagood JS, Owen CA. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2015;53(5):585-600. https://doi.org/10.1165/rcmb.2015-0020TR
33. Pilling D, Gomer RH. The development of serum amyloid P as a possible therapeutic. Front Immunol 2018;9:2328. https://doi.org/10.3389/fimmu.2018.02328
34. Perrot CY, Karampitsakos T, Herazo-Maya JD. Monocytes and macrophages: emerging mechanisms and novel therapeutic targets in pulmonary fibrosis. Am J Physiol Cell Physiol 2023;325(4):C1046-57. https://doi.org/10.1152/ajpcell.00302.2023
35. Fraser E, Denney L, Antanaviciute A, Blirando K, Vuppusetty C, Zheng Y et al. Multi-modal characterization of monocytes in idiopathic pulmonary fibrosis reveals a primed type I interferon immune phenotype. Front Immunol 2021;12:623430. https://doi.org/10.3389/fimmu.2021.623430
36. Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med 2011;184(5):569-81. https://doi.org/10.1164/rccm.201010-1719OC
37. Zielonka TM, Zycinska K, Radzikowska E, Filewska M, Bialas B, Obrowski MH et al. Influence of sera from interstitial lung disease patients on angiogenic activity of mononuclear cells. Adv Exp Med Biol 2013;756:139-45. https://doi.org/10.1007/978-94-007-4549-0_18
38. Hult EM, Gurczynski SJ, O'Dwyer DN, Zemans RL, Rasky A, Wang Y et al. Myeloid- and epithelial-derived heparin-binding epidermal growth factor-like growth factor promotes pulmonary fibrosis. Am J Respir Cell Mol Biol 2022;67(6):641-53. https://doi.org/10.1165/rcmb.2022-0174OC
39. Rojas J, Salazar J, Martínez MS, Palmar J, Bautista J, Chávez-Castillo M et al. Macrophage heterogeneity and plasticity: impact of macrophage biomarkers on atherosclerosis. Scientifica (Cairo) 2015;2015:851252. https://doi.org/10.1155/2015/851252
40. Cao Y, Rudrakshala J, Williams R, Rodriguez S, Sorkhdini P, Yang AX et al. CRTH2 mediates profibrotic macrophage differentiation and promotes lung fibrosis. Am J Respir Cell Mol Biol 2022;67(2):201-14. https://doi.org/10.1165/rcmb.2021-0504OC
41. Larson-Casey JL, Saleem K, Surolia R, Pandey J, Mack M, Antony VB et al. Myeloid heterogeneity mediates acute exacerbations of pulmonary fibrosis. J Immunol 2023;211(11):1714-24. https://doi.org/10.4049/jimmunol.2300053
42. Joshi N, Watanabe S, Verma R, Jablonski RP, Chen CI, Cheresh P et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur Respir J 2020;55(1):1900646. https://doi.org/10.1183/13993003.00646-2019
43. Akagawa KS. Functional heterogeneity of colony-stimulating factor-induced human monocyte-derived macrophages. Int J Hematol 2002;76(1):27-34. https://doi.org/10.1007/BF02982715
44. Huang WC, Sala-Newby GB, Susana A, Johnson JL, Newby AC. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-κB. PLoS One 2012;7(8):e42507. https://doi.org/10.1371/journal.pone.0042507
45. Pardo A, Selman M. Role of matrix metalloproteases in idiopathic pulmonary fibrosis. Fibrogenesis Tissue Repair 2012;5(Suppl 1):S9. https://doi.org/10.1186/1755-1536-5-S1-S9
46. Lin C, Rezaee F, Waasdorp M, Shi K, van der Poll T, Borensztajn K et al. Protease activated receptor-1 regulates macrophage-mediated cellular senescence: a risk for idiopathic pulmonary fibrosis. Oncotarget 2015;6(34):35304-14. https://doi.org/10.18632/oncotarget.6095
47. Zhang X, Ren Y, Xie B, Ye Q, Ban C, Zhang S et al. Blood monocyte counts as a prognostic biomarker and predictor in Chinese patients with idiopathic pulmonary fibrosis. Front Med (Lausanne) 2022;9:955125. https://doi.org/10.3389/fmed.2022.955125
48. Kreuter M, Lee JS, Tzouvelekis A, Oldham JM, Molyneaux PL, Weycker D et al. Monocyte count as a prognostic biomarker in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2021;204(1):74-81. https://doi.org/10.1164/rccm.202003-0669OC
49. Karampitsakos T, Torrisi S, Antoniou K, Manali E, Korbila I, Papaioannou O et al. Increased monocyte count and red cell distribution width as prognostic biomarkers in patients with idiopathic pulmonary fibrosis. Respir Res 2021;22(1):140. https://doi.org/10.1186/s12931-021-01725-9
50. Kreuter M, Lee JS, Tzouvelekis A, Oldham JM, Molyneaux PL, Weycker D et al. Modified blood cell GAP model as a prognostic biomarker in idiopathic pulmonary fibrosis. ERJ Open Res 2024;10(4):00666-2023. https://doi.org/10.1183/23120541.00666-2023
51. Coelho DJAB, Sousa C, Jacob M, Novais-Bastos H, Melo N, Mota PC et al. The role of monocyte count on monitoring patients with idiopathic pulmonary fibrosis under antifibrotic treatment. Eur Respir J 2020;56(suppl 64):722. https://doi.org/10.1183/13993003.congress-2020.722
52. Scott MKD, Quinn K, Li Q, Carroll R, Warsinske H, Vallania F et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: a retrospective, multicentre cohort study. Lancet Respir Med 2019;7(6):497-508. https://doi.org/10.1016/S2213-2600(18)30508-3
53. Bernardinello N, Grisostomi G, Cocconcelli E, Castelli G, Petrarulo S, Biondini D et al. The clinical relevance of lymphocyte to monocyte ratio in patients with Idiopathic Pulmonary Fibrosis (IPF). Respir Med 2022;191:106686. https://doi.org/10.1016/j.rmed.2021.106686
54. Kawamura K, Ichikado K, Anan K, Yasuda Y, Sekido Y, Suga M et al. Monocyte count and the risk for acute exacerbation of fibrosing interstitial lung disease: A retrospective cohort study. Chron Respir Dis 2020;17:1479973120909840. https://doi.org/10.1177/1479973120909840
55. Tsuneyoshi S, Zaizen Y, Tominaga M, Matama G, Umemoto S, Ohno S et al. Clinical significance of high monocyte counts for the continuous treatment with nintedanib. BMC Pulm Med 2023;23(1):242. https://doi.org/10.1186/s12890-023-02536-y
56. Perrot CY, Karampitsakos T, Herazo-Maya JD. Monocytes and macrophages: emerging mechanisms and novel therapeutic targets in pulmonary fibrosis. Am J Physiol Cell Physiol 2023;325(4):C1046-C1057. https://doi.org/10.1152/ajpcell.00302.2023
57. Achaiah A, Rathnapala A, Pereira A, Bothwell H, Dwivedi K, Barker R et al. Monocyte and neutrophil levels are potentially linked to progression to IPF for patients with indeterminate UIP CT pattern. BMJ Open Respir Res 2021;8(1):e000899. https://doi.org/10.1136/bmjresp-2021-000899
58. Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001;69(3):89-95. https://doi.org/10.1067/mcp.2001
 Esta revista está bajo una licencia de Creative Commons Attribution 4.0 International
Esta revista está bajo una licencia de Creative Commons Attribution 4.0 International