
CASO CLÍNICO | Respirar, 2025; 17(2): 201-206 | ISSN 2953-3414 | https://doi.org/10.55720/respirar.17.2.11
Analía Cánepa
https://orcid.org/0000-0002-9275-6467
Santiago Moyano
https://orcid.org/0000-0003-4200-3480
Florencia Artola
https://orcid.org/0000-0003-1574-6238
Martín Rebella
https://orcid.org/0000-0002-1925-4022
Manuel Baz
https://orcid.org/0000-0003-0226-1653
Recibido:
23 diciembre 2024
Aprobado:
28 marzo 2025

Esta revista está bajo una licencia de
Creative Commons Reconocimiento 4.0 Internacional.
Enfermedad pulmonar intersticial fibrosante en el síndrome antisintetasa: reporte de un caso clínico
Pulmonary Fibrosis and the Antisynthetase Syndrome: a Case Report
Analía Cánepa1 , Santiago Moyano1, Florencia Artola2, Martin Rebella1,3, Manuel Baz1
, Santiago Moyano1, Florencia Artola2, Martin Rebella1,3, Manuel Baz1
1. Hospital de Clínicas, Clínica Médica “C”, Montevideo, Uruguay.
2. Hospital de Clínicas, Cátedra de Neumología, Montevideo, Uruguay.
3. Hospital de Clínicas, Unidad de Enfermedades Autoinmunes Sistémicas, Montevideo, Uruguay..
Autor CORRESPONSAL:
Analía Cánepa, analia1521@hotmail.com
Resumen
El síndrome antisintetasa es una enfermedad poco frecuente que suele cursar con distintos grados de compromiso muscular, cutáneo, articular y pulmonar en función del perfil de autoanticuerpos involucrados en su patogénesis. La afectación pulmonar puede ser grave y tener un patrón fibrosante progresivo con importantes implicancias pronósticas y terapéuticas por lo que es fundamental su detección precoz mediante un screening oportuno basado en la tomografía de tórax de alta resolución y el estudio funcional respiratorio. Se presenta el caso clínico de una paciente con diagnóstico de miopatía inflamatoria anti Jo1 positiva refractaria a múltiples tratamientos inmunosupresores que instala en forma subaguda insuficiencia respiratoria, se llega al diagnóstico de enfermedad pulmonar intersticial fibrosante y presenta buena respuesta al tratamiento con nintedanib.
Palabras clave: miopatía inflamatoria, síndrome antisintetasa, fibrosis pulmonar.
Abstract
Antisynthetase syndrome is a rare disease that usually presents with different degrees of muscle, skin, joint and lung involvement depending on the profile of autoantibodies involved in its pathogenesis. Lung involvement can be serious and have a progressive fibrosing pattern with important prognostic and therapeutic implications, so its early detection through timely screening based on high-resolution chest tomography and respiratory functional study is essential. The clinical case of a patient with a diagnosis of anti-Jo1 positive inflammatory myopathy refractory to multiple immunosuppressive treatments that establish subacute respiratory failure is presented, leading to the diagnosis of fibrosing interstitial lung disease and presenting a good response to treatment with nintedanib.
Keywords: inflammatory myopathy, antisynthetase syndrome, pulmonary fibrosis.
Introducción
El síndrome antisintetasa se incluye dentro de las miopatías inflamatorias idiopáticas, un grupo de enfermedades autoinmunes que comparten algunas manifestaciones clínicas como inflamación muscular, erupción cutánea, artritis y alta prevalencia de enfermedad pulmonar intersticial (EPI).1 Los diferentes fenotipos entre las miopatías inflamatorias idiopáticas están estrechamente relacionados con el subtipo de autoanticuerpos específicos de miositis (MSA) y asociados a miositis (MAA)2 con implicancias en el diagnóstico, el curso de la enfermedad y la respuesta al tratamiento. La presencia de anticuerpos antisintetasa se asocia a una mayor frecuencia de EPI y peor pronóstico, con implicancias terapéuticas.1 (Tabla 1). El compromiso pulmonar suele ser de curso subagudo – crónico, puede preceder a la miopatía, ocurrir en forma concomitante o presentarse de manera aislada. Se presenta a continuación el caso clínico de una paciente con diagnóstico de miopatía inflamatoria refractaria anti-Jo1 positiva, en quien la afectación pulmonar se manifiesta 7 años después del inicio del compromiso muscular.
Tabla 1.
Anticuerpos antisintetasa
Adaptado de: “Tratado SER de Diagnóstico y Tratamiento de Enfermedades Autoinmunes Sistémicas”, Sociedad Española de Reumatología, 2018
|
Anticuerpos |
Frecuencia |
Asociaciones |
|
Anti-Jo 1 |
20-25% |
Enfermedad clásica con EPI, miositis, artritis y manos de mecánico. |
|
Anti-PL 7 |
5% |
Mayor incidencia y gravedad de la EPI. Compromiso esofágico y fenómeno de Raynaud. Escasa afectación muscular / articular. |
|
Anti-PL 12 |
5% |
|
|
Anti-EJ |
< 5% |
Enfermedad clásica con EPI, miositis y artritis. |
|
Anti-OJ |
< 5% |
Elevada frecuencia de EPI, en algunos casos presentan miositis, artritis y fenómeno de Raynaud. Buena respuesta a corticoides. |
|
Anti-KS |
< 1% |
Elevada frecuencia de EPI, algunos pacientes presentan miositis, artritis y fenómeno de Raynaud. |
|
Anti-ZO |
< 1% |
Variante de enfermedad clásica. |
|
Anti-YRS/Ha |
< 1% |
Variante de enfermedad clásica. |
Caso clínico
Se trata de una paciente de sexo femenino de 34 años, portadora de una miopatía inflamatoria anti-Jo 1 positiva diagnosticada en 2017, con compromiso exclusivamente muscular con afectación de cinturas escapular y pelviana. Se ha comportado como refractaria a múltiples líneas terapéuticas incluyendo corticoides, azatioprina, micofenolato de mofetilo y ciclosporina. Ha logrado un control parcial de la actividad muscular con rituximab semestral e inmunoglobulina intravenosa.
Recientemente, en una consulta ambulatoria refiere disnea de esfuerzo progresiva mMRC II – III de aproximadamente 3 meses de evolución, sin otros síntomas de la esfera respiratoria, en apirexia.
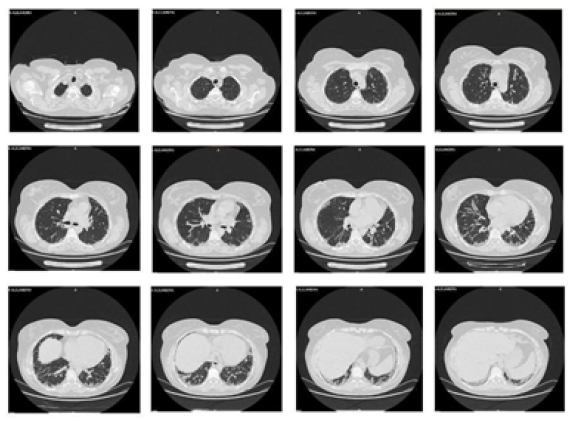
Del examen físico se destaca: facies cushingoide, sin lesiones en piel. Eupneica en reposo, sin taquicardia. Estertores crepitantes gruesos en ambas bases. Oximetría de pulso en reposo normal y, al esfuerzo, 88%. Entonces, es referida al Servicio de Emergencia para estudio, se le realiza analítica de laboratorio (Tabla 2) y radiografía de tórax (Figura 1). Se complementa la valoración con tomografía de tórax de alta resolución (Figura ٢) que identifica tractos densos biapicales; patrón reticular bilateral y simétrico de predominio subpleural basal y periférico con un gradiente apico-caudal; asocia áreas de panalización configurando un patrón de neumonía intersticial usual (NIU).
Tabla 2.
Bioquímica básica
|
Hemoglobina (Hb) |
10,6 g/dl |
|
Volumen corpuscular medio |
86 fL |
|
Hb corpuscular media |
28 pg |
|
Leucocitos |
18.420/mm3 |
|
Neutrófilos |
17.130/mm3 |
|
Linfocitos |
830/mm3 |
|
Plaquetas |
371.000/mm3 |
|
Velocidad de eritrosedimentación |
30 mm/h |
|
Creatininemia |
0,41 mg/dl |
|
Azoemia |
27 mg/dl |
|
Natremia |
136 mEq/l |
|
Kalemia |
4,8 mEq/l |
|
Creatinfosfoquinasa |
104 U/l |
|
Albuminemia |
4,20 g/dl |
Figura 1.
Radiografía de tórax frente y perfil que evidencia patrón retículo-nodular bilateral a franco predominio basal
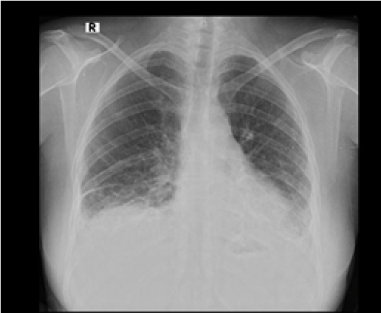
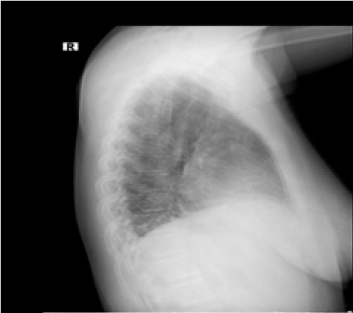
Figura 2.
Tomografía de tórax de alta resolución.
Asimismo, y dada su condición de inmunosupresión farmacológica, se realiza estudio microbiológico completo en vistas a descartar diagnóstico alternativo de infección respiratoria baja. Se destacan los siguientes resultados: antígeno neumocócico en orina negativo, PCR para virus respiratorios (virus respiratorio sincicial, COVID-19, influenza A y B) negativo. Hemocultivos negativos. Se estudia, además, mediante fibrobroncoscopia con lavado bronquio alveolar que muestra un citograma compuesto por abundantes macrófagos y leucocitos, 46% de eosinófilos, 33% de polimorfonucleares y 20% de linfocitos. Estudio bacteriológico directo y cultivo sin bacterias. Baciloscopias, cultivo y gene xpert negativo para Mycobacterium tuberculosis. Antígeno galactomanano negativo. Tinta china y cultivo micológico negativos. Investigación Pneumocystis jirovecii negativo.
Con diagnóstico de enfermedad pulmonar intersticial difusa vinculada al síndrome antisintetasa, se realiza exploración funcional respiratoria completa, con espirometría, volúmenes pulmonares, DLCO y fuerzas musculares, que evidencia una alteración restrictiva moderada a severa (CVF 1,79 L, 52%), una DLCO disminuida en grado severo (28 ml/mmHg/min) y fuerzas musculares disminuidas en grado severo (presión inspiratoria máxima 65 cmH2O, 75%, presión espiratoria máxima 19 cmH2O, 12%). Adicionalmente, presenta una distancia recorrida en 6 minutos (DR 6 min) de 360 m (54% del predicto) con saturación de oxígeno de pulso en reposo normal y desaturación de oxígeno durante la marcha de grado severo (88% a los 4 minutos de iniciada la marcha).
Se realiza, además, un ecocardiograma transtorácico sin elementos sugestivos de hipertensión pulmonar secundaria.
Dado el predominio de elementos fibróticos en la imagen pulmonar, se inicia tratamiento con nintedanib a dosis de 150 mg cada 12 h con buena tolerancia y mejoría sintomática y en los parámetros de función pulmonar a la semana 16 y 32 (Tabla 3).
Tabla 3.
Estudio funcional respiratorio
*Dudas técnicas de si realizó efectivamente el máximo esfuerzo.
|
Basal |
16 semanas |
32 semanas |
|
|
CVF (L) / (%) |
1,79 / 52 |
2,16 / 61 |
1,63 / 49%* |
|
DLCO (ml/mmHg/min) (%) |
7,3 / 28 |
7,9 / 33 |
No disponible |
|
DR 6 min (m/%) BORG inicial BORG final |
360 / 54 |
No disponible
|
430 / 66 |
|
0/6 |
0/6 |
||
|
4/6 |
0/6 |
Discusión
El síndrome antisintetasa es una enfermedad poco frecuente incluida dentro de las miopatías inflamatorias y se caracteriza por cursar con distintos grados de compromiso muscular, pulmonar, articular y cutáneo. Para su diagnóstico es importante la presencia de al menos un autoanticuerpo dirigido contra las ARN-t sintetasas de los cuales se conocen al menos 8, siendo el anti-Jo1 el más representativo.3,4 La triada clásica de síntomas descritos en el síndrome antisintetasa incluye enfermedad pulmonar intersticial, miositis y artritis. Sin embargo, y coincidente con el caso clínico presentado, solo una minoría de los pacientes exhiben la triada completa al inicio de la enfermedad.5
El compromiso muscular clínicamente se manifiesta por la presencia de debilidad muscular progresiva a nivel de cinturas escapular y pelviana, las manifestaciones articulares se expresan como una poliartritis bilateral, simétrica y no erosiva; en tanto que las lesiones cutáneas características son las “manos de mecánico” y más recientemente se han descrito lesiones similares a nivel de los pies conocidas como “pies de excursionista”.5 Además, pueden presentarse lesiones cutáneas características de dermatomiositis y el fenómeno de Raynaud que se encuentra en más de la mitad de los pacientes.
La afectación pulmonar tiene una prevalencia variable en las distintas series entre 64-98% y la gama de manifestaciones clínicas puede ir desde tos seca persistente hasta insuficiencia respiratoria severa. El patrón tomográfico se presenta como neumonía intersticial usual (NIU), neumonía intersticial organizada (NIO) o neumonía intersticial no específica (NINE) cuya respuesta terapéutica varía en función del grado de fibrosis. Al respecto, en 2023 Zannata et al. realizaron un estudio de cohorte multicéntrica cuyo objetivo fue investigar la aparición de EPI fibrosante progresiva en miopatías inflamatorias idiopáticas y los factores que potencialmente predicen este fenotipo. De los 253 pacientes analizados, 49% tenían EPI, 78% al momento del diagnóstico de la miopatía y 22% durante el seguimiento. El patrón predominante en la TACAR fue NINE (50%), seguido de NIU (28%) y NIO (22%), y el análisis multivariado identificó la presencia de anticuerpos anti-Jo1, anti-MDA5, anti-Ro52, un alto índice de actividad muscular, las manos de mecánico y el fenómeno de Raynaud como asociados de forma independiente con EPI. Al año de seguimiento, los pacientes con EPI mostraban un patrón fibrosante.6
En el caso de nuestra paciente, el compromiso que durante los primeros 7 años de evolución de su enfermedad fue exclusivamente muscular, refractario a múltiples líneas terapéuticas; presenta un mejor control de síntomas con rituximab e inmunoglobulina. La afectación pulmonar intersticial aparece una vez logrado el control de la actividad muscular y con predominio de elementos fibróticos en la TACAR por lo cual se define iniciar tratamiento con nintedanib. Se trata de un fármaco inhibidor de tirosinquinasas intracelulares que inhibe los procesos implicados en la progresión de la fibrosis pulmonar. Al respecto, el estudio INBUILD evaluó la eficacia y seguridad del nintedanib frente a placebo en 663 pacientes con EPI fibrosante progresiva de los cuales 25,6% tenían enfermedad autoinmune. Los pacientes tratados con nintedanib tuvieron una tasa de progresión de la EPI más lenta, independientemente del patrón en la TACAR, redujo de forma significativa la tasa de deterioro de la capacidad vital forzada (CVF) en la semana 52 de tratamiento.7 Nuestra paciente es concordante con los resultados de la literatura, en el sentido de no progresión, y muestra inclusive mejoría en parámetros de la funcionalidad respiratoria.
Conclusiones
El caso presentado pone en evidencia la importancia de realizar en forma rutinaria el screening de enfermedad pulmonar intersticial en pacientes con síndrome antisintetasa por su implicancia pronóstica y terapéutica. El uso de nintedanib en los pacientes con fenotipo fibrosante como el de nuestra paciente ofrece un escenario prometedor en términos de reducción de la tasa de progresión.
Financiamiento: los autores declaran que el trabajo no tuvo financiamiento.
Conflictos de interés: los autores declaran que no tienen conflictos de intereses relacionados con el tema de esta publicación.
Contribuciones de los autores: AC: conceptualización, curación de datos, investigación, administración de proyecto, recursos, visualización, escritura borrador original, redacción, revisión y edición. SM: curación de datos, recursos, visualización.FA: curación de datos, recursos, visualización, escritura del borrador original. MR: conceptualización, supervisión. MB: conceptualización, administración de proyecto, supervisión, validación, visualización, escritura del borrador original, redacción, revisión y edición.
El Editor en Jefe, Dr. Carlos Luna, realizó el seguimiento del proceso de revisión y aprobó este artículo.
Referencias